2022
Mathé-Hubert H; Amie R; Martin M; Gaffé J; Schneider D
Evolution of bacterial persistence to antibiotics during a 50,000-generation experiment in an antibiotic-free environment Journal Article
Antibiotics, 11 (4), pp. 451, 2022, ISSN: 2079-6382.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Genotypes and Phenotypes
@article{mathe-hubert2022,
title = {Evolution of bacterial persistence to antibiotics during a 50,000-generation experiment in an antibiotic-free environment},
author = {Hugo Mathé-Hubert and Rafika Amie and Mikaël Martin and Joël Gaffé and Dominique Schneider},
url = {https://www.mdpi.com/2079-6382/11/4/451},
doi = {10.3390/antibiotics11040451},
issn = {2079-6382},
year = {2022},
date = {2022-03-27},
urldate = {2022-03-27},
journal = {Antibiotics},
volume = {11},
number = {4},
pages = {451},
abstract = {Failure of antibiotic therapies causes >700,000 deaths yearly and involves both bacterial resistance and persistence. Persistence results in the relapse of infections by producing a tiny fraction of pathogen survivors that stay dormant during antibiotic exposure. From an evolutionary perspective, persistence is either a ‘bet-hedging strategy’ that helps to cope with stochastically changing environments or an unavoidable minimal rate of ‘cellular errors’ that lock the cells in a low activity state. Here, we analyzed the evolution of persistence over 50,000 bacterial generations in a stable environment by improving a published method that estimates the number of persister cells based on the growth of the reviving population. Our results challenged our understanding of the factors underlying persistence evolution. In one case, we observed a substantial decrease in persistence proportion, suggesting that the naturally observed persistence level is not an unavoidable minimal rate of ‘cellular errors’. However, although there was no obvious environmental stochasticity, in 11 of the 12 investigated populations, the persistence level was maintained during 50,000 bacterial generations.},
keywords = {Correlated Responses, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Jordan J A; Lenski R E; Card K J
Idiosyncratic fitness costs of ampicillin-resistant mutants derived from a long-term experiment with Escherichia coli Journal Article
Antibiotics, 11 (3), pp. 347, 2022, ISSN: 2079-6382.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution, Historical Contingency
@article{Jordan2022.02.06.479266,
title = {Idiosyncratic fitness costs of ampicillin-resistant mutants derived from a long-term experiment with \textit{Escherichia coli}},
author = {Jalin A. Jordan and Richard E. Lenski and Kyle J. Card},
url = {https://www.biorxiv.org/content/10.1101/2022.02.06.479266v1},
doi = {10.3390/antibiotics11030347},
issn = {2079-6382},
year = {2022},
date = {2022-03-13},
urldate = {2022-03-13},
journal = {Antibiotics},
volume = {11},
number = {3},
pages = {347},
publisher = {Cold Spring Harbor Laboratory},
abstract = {Antibiotic resistance is a growing concern that has prompted a renewed focus on drug discovery, stewardship, and evolutionary studies of the patterns and processes that underlie this phenomenon. A resistant strain’s competitive fitness relative to its sensitive counterparts in the absence of drug can impact its spread and persistence in both clinical and community settings. In a prior study, we examined the fitness of tetracycline-resistant clones that evolved from five different Escherichia coli genotypes, which had diverged during a long-term evolution experiment. In this study, we build on that work to examine whether ampicillin-resistant mutants are also less fit in the absence of the drug than their sensitive parents, and whether the cost of resistance is constant or variable among independently derived lines. Like the tetracycline-resistant lines, the ampicillin-resistant mutants were often less fit than their sensitive parents, with significant variation in the fitness costs among the mutants. This variation was not associated with the level of resistance conferred by the mutations, nor did it vary across the different parental backgrounds. In our earlier study, some of the variation in fitness costs associated with tetracycline resistance was explained by the effects of different mutations affecting the same cellular pathway and even the same gene. In contrast, the variance among the ampicillin-resistant mutants was associated with different sets of target genes. About half of the resistant clones suffered large fitness deficits, and their mutations impacted major outer-membrane proteins or subunits of RNA polymerases. The other mutants experienced little or no fitness costs and with, one exception, they had mutations affecting other genes and functions. Our findings underscore the importance of comparative studies on the evolution of antibiotic resistance, and they highlight the nuanced processes that shape these phenotypes.},
keywords = {Descendant Experiments, Genome Evolution, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
2021
Consuegra J; Gaffé J; Lenski R E; Hindré T; Barrick J E; Tenaillon O; Schneider D
Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria Journal Article
Nature Communications, 12 (1), pp. 980, 2021, ISSN: 2041-1723.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Mutation Rates
@article{Consuegra2021,
title = {Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria},
author = {Jessika Consuegra and Joël Gaffé and Richard E. Lenski and Thomas Hindré and Jeffrey E. Barrick and Olivier Tenaillon and Dominique Schneider},
url = {http://www.nature.com/articles/s41467-021-21210-7},
doi = {https://doi.org/10.1038/s41467-021-21210-7},
issn = {2041-1723},
year = {2021},
date = {2021-12-01},
urldate = {2021-12-01},
journal = {Nature Communications},
volume = {12},
number = {1},
pages = {980},
publisher = {Springer US},
abstract = {Insertion sequences (IS) are ubiquitous bacterial mobile genetic elements, and the mutations they cause can be deleterious, neutral, or beneficial. The long-term dynamics of IS elements and their effects on bacteria are poorly understood, including whether they are primarily genomic parasites or important drivers of adaptation by natural selection. Here, we investigate the dynamics of IS elements and their contribution to genomic evolution and fitness during a long-term experiment with \textit{Escherichia coli}. IS elements account for ~35% of the mutations that reached high frequency through 50,000 generations in those populations that retained the ancestral point-mutation rate. In mutator populations, IS-mediated mutations are only half as frequent in absolute numbers. In one population, an exceptionally high ~8-fold increase in IS 150 copy number is associated with the beneficial effects of early insertion mutations; however, this expansion later slowed down owing to reduced IS 150 activity. This population also achieves the lowest fitness, suggesting that some avenues for further adaptation are precluded by the IS 150 -mediated mutations. More generally, across all populations, we find that higher IS activity becomes detrimental to adaptation over evolutionary time. Therefore, IS-mediated mutations can both promote and constrain evolvability.},
keywords = {Fitness Trajectories, Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
Deatherage D E; Barrick J E
High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing Journal Article
Cell Systems, 12 (12), pp. 1187-1200, 2021, ISSN: 24054712.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous
@article{DEATHERAGE2021,
title = {High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing},
author = {Daniel E. Deatherage and Jeffrey E. Barrick},
url = {https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8678185/},
doi = {10.1016/j.cels.2021.08.011},
issn = {24054712},
year = {2021},
date = {2021-09-01},
urldate = {2021-09-01},
journal = {Cell Systems},
volume = {12},
number = {12},
pages = {1187-1200},
abstract = {Understanding how cells are likely to evolve can guide medical interventions and bioengineering efforts that must contend with unwanted mutations. The adaptome of a cell—the neighborhood of genetic changes that are most likely to drive adaptation in a given environment—can be mapped by tracking rare beneficial variants during the early stages of clonal evolution. We used multiplex adaptome capture sequencing (mAdCap-seq), a procedure that combines unique molecular identifiers and hybridization-based enrichment, to characterize mutations in eight \textit{Escherichia coli} genes known to be under selection in a laboratory environment. We tracked 301 mutations at frequencies as low as 0.01% and inferred the fitness effects of 240 of these mutations. There were distinct molecular signatures of selection on protein structure and function for the three genes with the most beneficial mutations. Our results demonstrate how mAdCap-seq can be used to deeply profile a targeted portion of a cell's adaptome.},
keywords = {Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
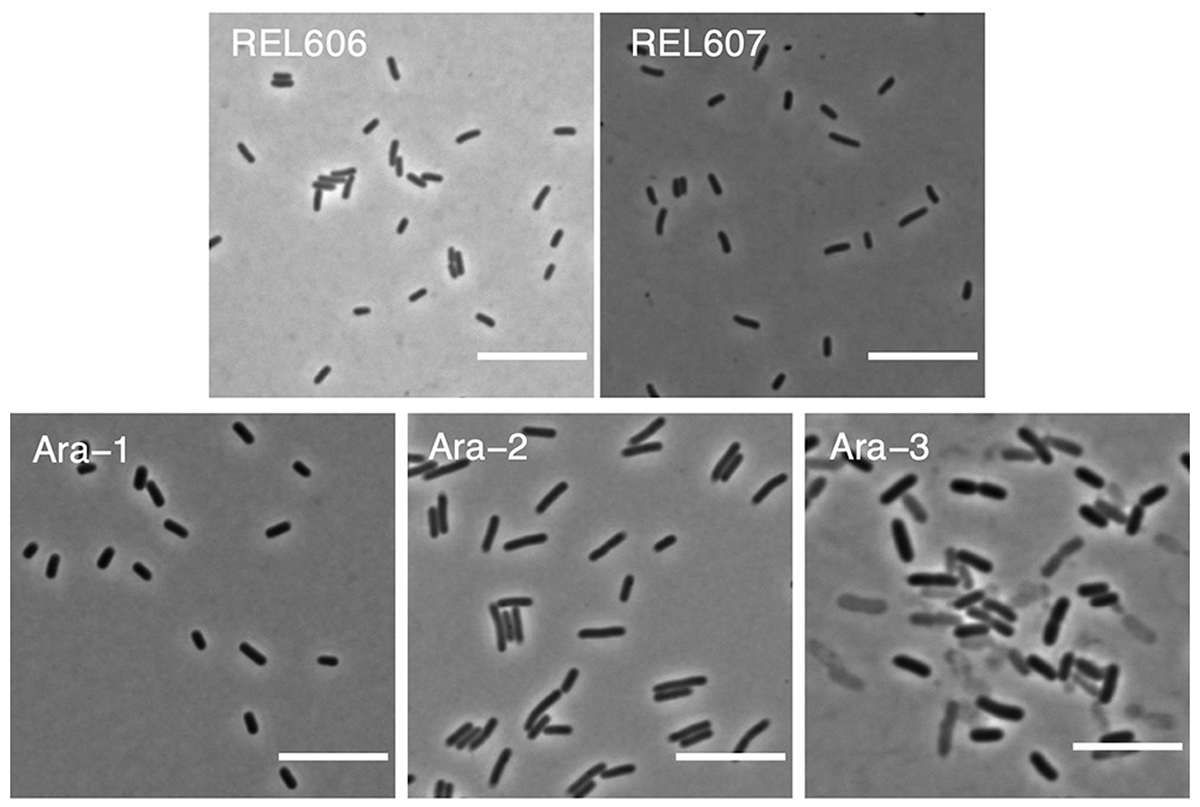
van Raay K; Stolyar S; Sevigny J; Draghi J; Lenski R E; Marx C J; Kerr B; Zaman L
Evolution with private resources reverses some changes from long-term evolution with public resources Unpublished
bioRxiv, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous
@unpublished{Raay2021,
title = {Evolution with private resources reverses some changes from long-term evolution with public resources},
author = {Katrina {van Raay} and Sergey Stolyar and Jordana Sevigny and Jeremy Draghi and Richard E. Lenski and Christopher J. Marx and Benjamin Kerr and Luis Zaman},
url = {https://www.biorxiv.org/content/10.1101/2021.07.11.451942v1},
doi = {https://doi.org/10.1101/2021.07.11.451942},
year = {2021},
date = {2021-07-12},
urldate = {2021-07-12},
journal = {bioRxiv},
pages = {2021.07.11.451942},
abstract = {A population under selection to improve one trait may evolve a sub-optimal state for another trait due to tradeoffs and other evolutionary constraints. How this evolution affects the capacity of a population to adapt when conditions change to favor the second trait is an open question. We investigated this question using isolates from a lineage spanning 60,000 generations of the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}, where cells have access to a shared pool of resources, and have evolved increased competitive ability and a concomitant reduction in numerical yield. Using media-in oil emulsions we shifted the focus of selection to numerical yield, where cells grew in isolated patches with private resources. We found that the time spent evolving under shared resources did not affect the ability to re-evolve toward higher numerical yield. The evolution of numerical yield commonly occurred through mutations in the phosphoenolpyruvate phosphotransferase system. These mutants exhibit slower uptake of glucose, making them poorer competitors for public resources, and produce smaller cells that release less carbon as overflow metabolites. Our results demonstrate that mutations that were not part of adaptation under one selective regime may enable access to ancestral phenotypes when selection changes to favor evolutionary reversion. },
howpublished = {bioRxiv},
keywords = {Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {unpublished}
}
Grant N A; Maddamsetti R; Lenski R E
Maintenance of Metabolic Plasticity despite Relaxed Selection in a Long-Term Evolution Experiment with Escherichia coli Journal Article
The American Naturalist, 198 (1), pp. 93–112, 2021, ISSN: 0003-0147.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes
@article{Grant2021b,
title = {Maintenance of Metabolic Plasticity despite Relaxed Selection in a Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Nkrumah A. Grant and Rohan Maddamsetti and Richard E. Lenski},
url = {https://www.journals.uchicago.edu/doi/10.1086/714530},
doi = {10.1086/714530},
issn = {0003-0147},
year = {2021},
date = {2021-07-01},
urldate = {2021-07-01},
journal = {The American Naturalist},
volume = {198},
number = {1},
pages = {93--112},
abstract = {Traits that are unused in a given environment are subject to processes that tend to erode them, leading to reduced fitness in other environments. Although this general tendency is clear, we know much less about why some traits are lost while others are retained and about the roles of mutation and selection in generating different responses. We addressed these issues by examining populations of a facultative anaerobe, \textit{Escherichia coli}, that have evolved for 130 years in the presence of oxygen, with relaxed selection for anaerobic growth and the associated metabolic plasticity. We asked whether evolution led to the loss, improvement, or maintenance of anaerobic growth, and we analyzed gene expression and mutational data sets to understand the outcomes. We identified genomic signatures of both positive and purifying selection on aerobic-specific genes, while anaerobic-specific genes showed clear evidence of relaxed selection. We also found parallel evolution at two interacting loci that regulate anaerobic growth. We competed the ancestor and evolved clones from each population in an anoxic environment, and we found that anaerobic fitness had not decayed, despite relaxed selection. In summary, relaxed selection does not necessarily reduce an organism's fitness in other environments. Instead, the genetic architecture of the traits under relaxed selection and their correlations with traits under positive and purifying selection may sometimes determine evolutionary outcomes.},
keywords = {Correlated Responses, Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Karkare K; Lai H; Azevedo R B R; Cooper T F
Historical Contingency Causes Divergence in Adaptive Expression of the lac Operon Journal Article
Molecular Biology and Evolution, 38 (7), pp. 2869–2879, 2021, ISSN: 1537-1719.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genotypes and Phenotypes
@article{10.1093/molbev/msab077,
title = {Historical Contingency Causes Divergence in Adaptive Expression of the lac Operon},
author = {Kedar Karkare and Huei-Yi Lai and Ricardo B. R. Azevedo and Tim F. Cooper},
editor = {Patricia Wittkopp},
url = {https://academic.oup.com/mbe/article/38/7/2869/6179336},
doi = {10.1093/molbev/msab077},
issn = {1537-1719},
year = {2021},
date = {2021-06-01},
urldate = {2021-06-01},
journal = {Molecular Biology and Evolution},
volume = {38},
number = {7},
pages = {2869--2879},
abstract = {Populations of \textit{Escherichia coli} selected in constant and fluctuating environments containing lactose often adapt by substituting mutations in the lacI repressor that cause constitutive expression of the lac operon. These mutations occur at a high rate and provide a significant benefit. Despite this, eight of 24 populations evolved for 8,000 generations in environments containing lactose contained no detectable repressor mutations. We report here on the basis of this observation. We find that, given relevant mutation rates, repressor mutations are expected to have fixed in all evolved populations if they had maintained the same fitness effect they confer when introduced to the ancestor. In fact, reconstruction experiments demonstrate that repressor mutations have become neutral or deleterious in those populations in which they were not detectable. Populations not fixing repressor mutations nevertheless reached the same fitness as those that did fix them, indicating that they followed an alternative evolutionary path that made redundant the potential benefit of the repressor mutation, but involved unique mutations of equivalent benefit. We identify a mutation occurring in the promoter region of the uspB gene as a candidate for influencing the selective choice between these paths. Our results detail an example of historical contingency leading to divergent evolutionary outcomes.},
keywords = {Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Card K J; Jordan J A; Lenski R E
Idiosyncratic variation in the fitness costs of tetracycline-resistance mutations in Escherichia coli Journal Article
Evolution, 75 (5), pp. 1230-1238, 2021, ISSN: 0014-3820.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{nokey,
title = {Idiosyncratic variation in the fitness costs of tetracycline-resistance mutations in \textit{Escherichia coli}},
author = {Kyle J. Card and Jalin A. Jordan and Richard E. Lenski},
url = {https://onlinelibrary.wiley.com/doi/10.1111/evo.14203},
doi = {10.1111/evo.14203},
issn = {0014-3820},
year = {2021},
date = {2021-02-25},
urldate = {2021-02-25},
journal = {Evolution},
volume = {75},
number = {5},
pages = {1230-1238},
abstract = {A bacterium's fitness relative to its competitors, both in the presence and absence of antibiotics, plays a key role in its ecological success and clinical impact. In this study, we examine whether tetracycline-resistant mutants are less fit in the absence of the drug than their sensitive parents, and whether the fitness cost of resistance is constant or variable across independently derived lines. Tetracycline-resistant lines suffered, on average, a reduction in fitness of almost 8%. There was substantial among-line variation in the fitness cost. This variation was not associated with the level of resistance conferred by the mutations, nor did it vary significantly across several genetic backgrounds. The two resistant lines with the most extreme fitness costs involved functionally unrelated mutations on different genetic backgrounds. However, there was also significant variation in the fitness costs for mutations affecting the same pathway and even different alleles of the same gene. Our findings demonstrate that the fitness costs of antibiotic resistance do not always correlate with the phenotypic level of resistance or the underlying genetic changes. Instead, these costs reflect the idiosyncratic effects of particular resistance mutations and the genetic backgrounds in which they occur.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
Card K J; Thomas M D; Graves J L; Barrick J E; Lenski R E
Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with Escherichia coli Journal Article
Proceedings of the National Academy of Sciences of the United States of America, 118 (5), pp. e2016886118, 2021, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution, Historical Contingency
@article{Card2021,
title = {Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with \textit{Escherichia coli}},
author = {Kyle J. Card and Misty D. Thomas and Joseph L. Graves and Jeffrey E. Barrick and Richard E. Lenski},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.2016886118},
doi = {10.1073/pnas.2016886118},
issn = {0027-8424},
year = {2021},
date = {2021-02-01},
urldate = {2021-02-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {118},
number = {5},
pages = {e2016886118},
abstract = {Antibiotic resistance is a growing health concern. Efforts to control resistance would benefit from an improved ability to forecast when and how it will evolve. Epistatic interactions between mutations can promote divergent evolutionary trajectories, which complicates our ability to predict evolution. We recently showed that differences between genetic backgrounds can lead to idiosyncratic responses in the evolvability of phenotypic resistance, even among closely related \textit{Escherichia coli} strains. In this study, we examined whether a strain's genetic background also influences the genotypic evolution of resistance. Do lineages founded by different genotypes take parallel or divergent mutational paths to achieve their evolved resistance states? We addressed this question by sequencing the complete genomes of antibiotic-resistant clones that evolved from several different genetic starting points during our earlier experiments. We first validated our statistical approach by quantifying the specificity of genomic evolution with respect to antibiotic treatment. As expected, mutations in particular genes were strongly associated with each drug. Then, we determined that replicate lines evolved from the same founding genotypes had more parallel mutations at the gene level than lines evolved from different founding genotypes, although these effects were more subtle than those showing antibiotic specificity. Taken together with our previous work, we conclude that historical contingency can alter both genotypic and phenotypic pathways to antibiotic resistance.},
keywords = {Descendant Experiments, Genome Evolution, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Maddamsetti R
Selection Maintains Protein Interactome Resilience in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Genome Biology and Evolution, 13 (6), pp. evab074, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Genotypes and Phenotypes
@article{Maddamsetti2021,
title = {Selection Maintains Protein Interactome Resilience in the Long-Term Evolution Experiment with \emph{Escherichia coli}},
author = {Rohan Maddamsetti},
url = {https://academic.oup.com/gbe/article/13/6/evab074/6240992},
doi = {https://doi.org/10.1093/gbe/evab074},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {Genome Biology and Evolution},
volume = {13},
number = {6},
pages = {evab074},
abstract = {Most cellular functions are carried out by a dynamic network of interacting proteins. An open question is whether the network properties of protein interactomes represent phenotypes under natural selection. One proposal is that protein interactomes have evolved to be resilient, such that they tend to maintain connectivity when proteins are removed from the network. This hypothesis predicts that interactome resilience should be maintained by natural selection during long-term experimental evolution. I tested this prediction by modeling the evolution of protein–protein interaction (PPI) networks in Lenski’s long-term evolution experiment with \textit{Escherichia coli} (LTEE). In this test, I removed proteins affected by nonsense, insertion, deletion, and transposon mutations in evolved LTEE strains, and measured the resilience of the resulting networks. I compared the rate of change of network resilience in each LTEE population to the rate of change of network resilience for corresponding randomized networks. The evolved PPI networks are significantly more resilient than networks in which random proteins have been deleted. Moreover, the evolved networks are generally more resilient than networks in which the random deletion of proteins was restricted to those disrupted in LTEE. These results suggest that evolution in the LTEE has favored PPI networks that are, on average, more resilient than expected from the genetic variation across the evolved strains. My findings therefore support the hypothesis that selection maintains protein interactome resilience over evolutionary time.},
keywords = {Genome Evolution, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Maddamsetti R
Universal Constraints on Protein Evolution in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Genome Biology and Evolution, 13 (6), pp. evab070, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Genotypes and Phenotypes
@article{Maddamsetti2021b,
title = {Universal Constraints on Protein Evolution in the Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Rohan Maddamsetti},
url = {https://academic.oup.com/gbe/article/13/6/evab070/6226398},
doi = {https://doi.org/10.1093/gbe/evab070},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {Genome Biology and Evolution},
volume = {13},
number = {6},
pages = {evab070},
abstract = {Although it is well known that abundant proteins evolve slowly across the tree of life, there is little consensus for why this is true. Here, I report that abundant proteins evolve slowly in the hypermutator populations of Lenski’s long-term evolution experiment with \textit{Escherichia coli} (LTEE). Specifically, the density of all observed mutations per gene, as measured in metagenomic time series covering 60,000 generations of the LTEE, significantly anticorrelates with mRNA abundance, protein abundance, and degree of protein–protein interaction. The same pattern holds for nonsynonymous mutation density. However, synonymous mutation density, measured across the LTEE hypermutator populations, positively correlates with protein abundance. These results show that universal constraints on protein evolution are visible in data spanning three decades of experimental evolution. Therefore, it should be possible to design experiments to answer why abundant proteins evolve slowly.},
keywords = {Genome Evolution, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Grant N A; abdel Magid A; Franklin J; Dufour Y; Lenski R E
Changes in cell size and shape during 50,000 generations of experimental evolution with Escherichia coli Journal Article
Journal of Bacteriology, 203 (10), pp. e00469-20, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Genotypes and Phenotypes
@article{Grant2021,
title = {Changes in cell size and shape during 50,000 generations of experimental evolution with \textit{Escherichia coli}},
author = {Nkrumah A. Grant and Ali {abdel Magid} and Joshua Franklin and Yann Dufour and Richard E. Lenski},
url = {https://journals.asm.org/doi/10.1128/JB.00469-20},
doi = {10.1128/JB.00469-20},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {Journal of Bacteriology},
volume = {203},
number = {10},
pages = {e00469-20},
abstract = {Bacteria adopt a wide variety of sizes and shapes, with many species exhibiting stereotypical morphologies. How morphology changes, and over what timescales, is less clear. Previous work examining cell morphology in an experiment with \textit{Escherichia coli} showed that populations evolved larger cells and, in some cases, cells that were less rod-like. That experiment has now run for over two more decades. Meanwhile, genome sequence data are available for these populations, and new computational methods enable high-throughput microscopic analyses. In this study, we measured stationary-phase cell volumes for the ancestor and 12 populations at 2,000, 10,000, and 50,000 generations, including measurements during exponential growth at the last time point. We measured the distribution of cell volumes for each sample using a Coulter counter and microscopy, the latter of which also provided data on cell shape. Our data confirm the trend toward larger cells while also revealing substantial variation in size and shape across replicate populations. Most populations first evolved wider cells but later reverted to the ancestral length-to-width ratio. All but one population evolved mutations in rod shape maintenance genes. We also observed many ghost-like cells in the only population that evolved the novel ability to grow on citrate, supporting the hypothesis that this lineage struggles with maintaining balanced growth. Lastly, we show that cell size and fitness remain correlated across 50,000 generations. Our results suggest that larger cells are beneficial in the experimental environment, while the reversion toward ancestral length-to-width ratios suggests partial compensation for the less favorable surface area-to-volume ratios of the evolved cells.},
keywords = {Cell Morphology, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Gifford I; Dasgupta A; Barrick J E
Rates of gene conversions between Escherichia coli ribosomal operons Journal Article
G3: Genes, Genomes, Genetics, 11 (2), pp. jkaa002, 2021, ISSN: 21601836.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Mutation Rates
@article{Gifford2021,
title = {Rates of gene conversions between \emph{Escherichia coli} ribosomal operons},
author = {Isaac Gifford and Aurko Dasgupta and Jeffrey E. Barrick},
url = {https://academic.oup.com/g3journal/article/11/2/jkaa002/5974039},
doi = {10.1093/g3journal/jkaa002},
issn = {21601836},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {G3: Genes, Genomes, Genetics},
volume = {11},
number = {2},
pages = {jkaa002},
abstract = {Due to their universal presence and high sequence conservation, ribosomal RNA (rRNA) sequences are used widely in phylogenetics for inferring evolutionary relationships between microbes and in metagenomics for analyzing the composition of microbial communities. Most microbial genomes encode multiple copies of rRNA genes to supply cells with sufficient capacity for protein synthesis. These copies typically undergo concerted evolution that keeps their sequences identical, or nearly so, due to gene conversion, a type of intragenomic recombination that changes one copy of a homologous sequence to exactly match another. Widely varying rates of rRNA gene conversion have previously been estimated by comparative genomics methods and using genetic reporter assays. To more directly measure rates of rRNA intragenomic recombination, we sequenced the seven \textit{Escherichia coli} rRNA operons in 15 lineages that were evolved for ~13,750 generations with frequent single-cell bottlenecks that reduce the effects of selection. We identified 38 gene conversion events and estimated an overall rate of intragenomic recombination within the 16S and 23S genes between rRNA copies of 3.6 × 10^{−4} per genome per generation or 8.6 × 10^{6} per rRNA operon per homologous donor operon per generation. This rate varied only slightly from random expectations at different sites within the rRNA genes and between rRNA operons located at different positions in the genome. Our accurate estimate of the rate of rRNA gene conversions fills a gap in our quantitative understanding of how ribosomal sequences and other multicopy elements diversify and homogenize during microbial genome evolution.},
keywords = {Descendant Experiments, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2020
Atolia E; Cesar S; Arjes H A; Rajendram M; Shi H; Knapp B D; Khare S; Aranda-Díaz A; Lenski R E; Huang K C
Environmental and Physiological Factors Affecting High-Throughput Measurements of Bacterial Growth Journal Article
mBio, 11 (5), pp. 1–19, 2020, ISSN: 2161-2129.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology
@article{Atolia2020,
title = {Environmental and Physiological Factors Affecting High-Throughput Measurements of Bacterial Growth},
author = {Esha Atolia and Spencer Cesar and Heidi A Arjes and Manohary Rajendram and Handuo Shi and Benjamin D Knapp and Somya Khare and Andrés Aranda-Díaz and Richard E. Lenski and Kerwyn Casey Huang},
editor = {Kelly T. Hughes},
url = {https://journals.asm.org/doi/10.1128/mBio.01378-20},
doi = {10.1128/mBio.01378-20},
issn = {2161-2129},
year = {2020},
date = {2020-10-01},
urldate = {2020-10-01},
journal = {mBio},
volume = {11},
number = {5},
pages = {1--19},
abstract = {How starved bacteria adapt and multiply under replete nutrient conditions is intimately linked to their history of previous growth, their physiological state, and the surrounding environment. While automated equipment has enabled high-throughput growth measurements, data interpretation and knowledge gaps regarding the determinants of growth kinetics complicate comparisons between strains. Here, we present a framework for growth measurements that improves accuracy and attenuates the effects of growth history. We determined that background absorbance quantification and multiple passaging cycles allow for accurate growth rate measurements even in carbon-poor media, which we used to reveal growth-rate increases during long-term laboratory evolution of \textit{Escherichia coli}. Using mathematical modeling, we showed that maximum growth rate depends on initial cell density. Finally, we demonstrated that growth of Bacillus subtilis with glycerol inhibits the future growth of most of the population, due to lipoteichoic acid synthesis. These studies highlight the challenges of accurate quantification of bacterial growth behaviors.},
keywords = {Demography and Ecology},
pubstate = {published},
tppubtype = {article}
}
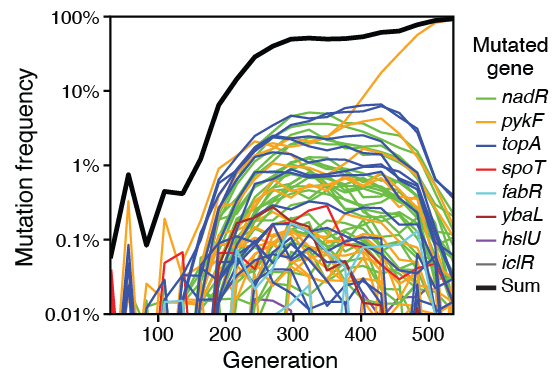
Maddamsetti R; Grant N A
Divergent Evolution of Mutation Rates and Biases in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Genome Biology and Evolution, 12 (9), pp. 1591-1603, 2020, ISSN: 1759-6653.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{nokey,
title = {Divergent Evolution of Mutation Rates and Biases in the Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Rohan Maddamsetti and Nkrumah A. Grant},
editor = {George Zhang},
url = {https://academic.oup.com/gbe/article/12/9/1591/5898197},
doi = {10.1093/gbe/evaa178},
issn = {1759-6653},
year = {2020},
date = {2020-08-21},
urldate = {2020-08-21},
journal = {Genome Biology and Evolution},
volume = {12},
number = {9},
pages = {1591-1603},
abstract = {All organisms encode enzymes that replicate, maintain, pack, recombine, and repair their genetic material. For this reason, mutation rates and biases also evolve by mutation, variation, and natural selection. By examining metagenomic time series of the Lenski long-term evolution experiment (LTEE) with \textit{Escherichia coli} (Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. 2017. The dynamics of molecular evolution over 60,000 generations. Nature 551(7678):45–50.), we find that local mutation rate variation has evolved during the LTEE. Each LTEE population has evolved idiosyncratic differences in their rates of point mutations, indels, and mobile element insertions, due to the fixation of various hypermutator and antimutator alleles. One LTEE population, called Ara+3, shows a strong, symmetric wave pattern in its density of point mutations, radiating from the origin of replication. This pattern is largely missing from the other LTEE populations, most of which evolved missense, indel, or structural mutations in \textit{topA}, \textit{fis}, and \textit{dusB}—loci that all affect DNA topology. The distribution of mutations in those genes over time suggests epistasis and historical contingency in the evolution of DNA topology, which may have in turn affected local mutation rates. Overall, the replicate populations of the LTEE have largely diverged in their mutation rates and biases, even though they have adapted to identical abiotic conditions.},
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
Blount Z D; Maddamsetti R; Grant N A; Ahmed S T; Jagdish T; Baxter J A; Sommerfeld B A; Tillman A; Moore J; Slonczewski J L; Barrick J E; Lenski R E
Genomic and phenotypic evolution of Escherichia coli in a novel citrate-only resource environment Journal Article
eLife, 9 , pp. 1–64, 2020, ISSN: 2050-084X.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes
@article{Blount2020,
title = {Genomic and phenotypic evolution of \textit{Escherichia coli} in a novel citrate-only resource environment},
author = {Zachary D. Blount and Rohan Maddamsetti and Nkrumah A. Grant and Sumaya T. Ahmed and Tanush Jagdish and Jessica A. Baxter and Brooke A. Sommerfeld and Alice Tillman and Jeremy Moore and Joan L. Slonczewski and Jeffrey E. Barrick and Richard E. Lenski},
url = {https://elifesciences.org/articles/55414},
doi = {10.7554/eLife.55414},
issn = {2050-084X},
year = {2020},
date = {2020-05-01},
urldate = {2020-05-01},
journal = {eLife},
volume = {9},
pages = {1--64},
abstract = {Evolutionary innovations allow populations to colonize new ecological niches. We previously reported that aerobic growth on citrate (Cit^{+}) evolved in an \textit{Escherichia coli} population during adaptation to a minimal glucose medium containing citrate (DM25). Cit^{+} variants can also grow in citrate-only medium (DM0), a novel environment for \textit{E. coli}. To study adaptation to this niche, we founded two sets of Cit^{+} populations and evolved them for 2500 generations in DM0 or DM25. The evolved lineages acquired numerous parallel mutations, many mediated by transposable elements. Several also evolved amplifications of regions containing the \textit{maeA} gene. Unexpectedly, some evolved populations and clones show apparent declines in fitness. We also found evidence of substantial cell death in Cit^{+} clones. Our results thus demonstrate rapid trait refinement and adaptation to the new citrate niche, while also suggesting a recalcitrant mismatch between \textit{E. coli} physiology and growth on citrate.},
keywords = {Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Barrick J E; Deatherage D E; Lenski R E
Banzhaf, Wolfgang; Cheng, Betty H C; Deb, Kalyanmoy; Holekamp, Kay E; Lenski, Richard E; Ofria, Charles; Pennock, Robert T; Punch, William F; Whittaker, Danielle J (Ed.): Evolution in Action: Past, Present and Future: A Festschrift in Honor of Erik D. Goodman, pp. 77–89, Springer International Publishing, Cham, 2020, ISBN: 978-3-030-39831-6.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories
@inbook{Barrick2020,
title = {A Test of the Repeatability of Measurements of Relative Fitness in the Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Jeffrey E. Barrick and Daniel E. Deatherage and Richard E. Lenski},
editor = {Wolfgang Banzhaf and Betty H C Cheng and Kalyanmoy Deb and Kay E Holekamp and Richard E Lenski and Charles Ofria and Robert T Pennock and William F Punch and Danielle J Whittaker},
url = {http://link.springer.com/10.1007/978-3-030-39831-6_8},
doi = {10.1007/978-3-030-39831-6_8},
isbn = {978-3-030-39831-6},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
booktitle = {Evolution in Action: Past, Present and Future: A Festschrift in Honor of Erik D. Goodman},
pages = {77--89},
publisher = {Springer International Publishing},
address = {Cham},
abstract = {Experimental studies of evolution using microbes have a long tradition, and these studies have increased greatly in number and scope in recent decades. Most such experiments have been short in duration, typically running for weeks or months. A venerable exception, the long-term evolution experiment (LTEE) with \textit{Escherichia coli} has continued for 30 years and 70,000 bacterial generations. The LTEE has become one of the cornerstones of the field of experimental evolution, in general, and the BEACON Center for the Study of Evolution in Action, in particular. Science laboratories and experiments usually have finite lifespans, but we hope that the LTEE can continue far into the future. There are practical issues associated with maintaining such a long-term experiment. One issue, which we address here, is whether key measurements made at one time and place are reproducible, within reasonable limits, at other times and places. This issue comes to the forefront when one considers moving an experiment like the LTEE from one lab to another. To that end, the Barrick lab at The University of Texas at Austin, measured the fitness values of samples from the 12 LTEE populations at 2,000, 10,000, and 50,000 generations and compared the new data to data previously obtained at Michigan State University. On balance, the datasets agree very well. More generally, this finding shows the value of simplicity in experimental design, such as using a chemically defined growth medium and appropriately storing samples from microbiological experiments. Even so, one must be vigilant in checking assumptions and procedures given the potential for uncontrolled factors (e.g., water quality) to affect outcomes. This vigilance is perhaps especially important for a trait like fitness, which integrates all aspects of organismal performance and may therefore be sensitive to any number of subtle environmental influences.},
keywords = {Fitness Trajectories},
pubstate = {published},
tppubtype = {inbook}
}
2019
Card K J; LaBar T; Gomez J B; Lenski R E
Historical contingency in the evolution of antibiotic resistance after decades of relaxed selection Journal Article
PLOS Biology, 17 (10), pp. e3000397, 2019, ISSN: 1545-7885.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Descendant Experiments, Historical Contingency
@article{nokey,
title = {Historical contingency in the evolution of antibiotic resistance after decades of relaxed selection},
author = {Kyle J. Card and Thomas LaBar and Jasper B. Gomez and Richard E. Lenski},
url = {https://dx.plos.org/10.1371/journal.pbio.3000397},
doi = {10.1371/journal.pbio.3000397},
issn = {1545-7885},
year = {2019},
date = {2019-10-23},
urldate = {2019-10-23},
journal = {PLOS Biology},
volume = {17},
number = {10},
pages = {e3000397},
abstract = {Populations often encounter changed environments that remove selection for the maintenance of particular phenotypic traits. The resulting genetic decay of those traits under relaxed selection reduces an organism’s fitness in its prior environment. However, whether and how such decay alters the subsequent evolvability of a population upon restoration of selection for a previously diminished trait is not well understood. We addressed this question using \textit{Escherichia coli} strains from the long-term evolution experiment (LTEE) that independently evolved for multiple decades in the absence of antibiotics. We first confirmed that these derived strains are typically more sensitive to various antibiotics than their common ancestor. We then subjected the ancestral and derived strains to various concentrations of these drugs to examine their potential to evolve increased resistance. We found that evolvability was idiosyncratic with respect to initial genotype; that is, the derived strains did not generally compensate for their greater susceptibility by “catching up” to the resistance level of the ancestor. Instead, the capacity to evolve increased resistance was constrained in some backgrounds, implying that evolvability depended upon prior mutations in a historically contingent fashion. We further subjected a time series of clones from one LTEE population to tetracycline and determined that an evolutionary constraint arose early in that population, corroborating the role of contingency. In summary, relaxed selection not only can drive populations to increased antibiotic susceptibility, but it can also affect the subsequent evolvability of antibiotic resistance in an unpredictable manner. This conclusion has potential implications for public health, and it underscores the need to consider the genetic context of pathogens when designing drug-treatment strategies.},
keywords = {Correlated Responses, Descendant Experiments, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Lamrabet O; Plumbridge J; Martin M; Lenski R E; Schneider D; Hindré T
Plasticity of Promoter-Core Sequences Allows Bacteria to Compensate for the Loss of a Key Global Regulatory Gene Journal Article
Molecular Biology and Evolution, 36 (6), pp. 1121–1133, 2019, ISSN: 0737-4038.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{Lamrabet2019b,
title = {Plasticity of Promoter-Core Sequences Allows Bacteria to Compensate for the Loss of a Key Global Regulatory Gene},
author = {Otmane Lamrabet and Jacqueline Plumbridge and Mikaël Martin and Richard E. Lenski and Dominique Schneider and Thomas Hindré},
editor = {Csaba Pal},
url = {https://academic.oup.com/mbe/article/36/6/1121/5368491},
doi = {10.1093/molbev/msz042},
issn = {0737-4038},
year = {2019},
date = {2019-06-01},
urldate = {2019-06-01},
journal = {Molecular Biology and Evolution},
volume = {36},
number = {6},
pages = {1121--1133},
abstract = {Transcription regulatory networks (TRNs) are of central importance for both short-term phenotypic adaptation in response to environmental fluctuations and long-term evolutionary adaptation, with global regulatory genes often being targets of natural selection in laboratory experiments. Here, we combined evolution experiments, whole-genome resequencing, and molecular genetics to investigate the driving forces, genetic constraints, and molecular mechanisms that dictate how bacteria can cope with a drastic perturbation of their TRNs. The \textit{crp} gene, encoding a major global regulator in \textit{Escherichia coli}, was deleted in four different genetic backgrounds, all derived from the Long-Term Evolution Experiment (LTEE) but with different TRN architectures. We confirmed that \textit{crp} deletion had a more deleterious effect on growth rate in the LTEE-adapted genotypes; and we showed that the \textit{ptsG} gene, which encodes the major glucose-PTS transporter, gained CRP (cyclic AMP receptor protein) dependence over time in the LTEE. We then further evolved the four \textit{crp}-deleted genotypes in glucose minimal medium, and we found that they all quickly recovered from their growth defects by increasing glucose uptake. We showed that this recovery was specific to the selective environment and consistently relied on mutations in the cis-regulatory region of \textit{ptsG}, regardless of the initial genotype. These mutations affected the interplay of transcription factors acting at the promoters, changed the intrinsic properties of the existing promoters, or produced new transcription initiation sites. Therefore, the plasticity of even a single promoter region can compensate by three different mechanisms for the loss of a key regulatory hub in the \textit{E. coli} TRN.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
Lamrabet O; Martin M; Lenski R E; Schneider D
Changes in Intrinsic Antibiotic Susceptibility during a Long-Term Evolution Experiment with Escherichia coli Journal Article
mBio, 10 (2), pp. 1–12, 2019, ISSN: 2161-2129.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses
@article{Lamrabet2019,
title = {Changes in Intrinsic Antibiotic Susceptibility during a Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Otmane Lamrabet and Mikaël Martin and Richard E. Lenski and Dominique Schneider},
editor = {Julian E. Davies},
url = {https://journals.asm.org/doi/10.1128/mBio.00189-19},
doi = {10.1128/mBio.00189-19},
issn = {2161-2129},
year = {2019},
date = {2019-04-01},
urldate = {2019-04-01},
journal = {mBio},
volume = {10},
number = {2},
pages = {1--12},
abstract = {Resistance to antibiotics often evolves when bacteria encounter antibiotics. However, bacterial strains and species without any known exposure to these drugs also vary in their intrinsic susceptibility. In many cases, evolved resistance has been shown to be costly to the bacteria, such that resistant types have reduced competitiveness relative to their sensitive progenitors in the absence of antibiotics. In this study, we examined changes in the susceptibilities of 12 populations of \textit{Escherichia coli} to 15 antibiotics after 2,000 and 50,000 generations without exposure to any drug. The evolved bacteria tended to become more susceptible to most antibiotics, with most of the change occurring during the first 2,000 generations, when the bacteria were undergoing rapid adaptation to their experimental conditions. On balance, our findings indicate that bacteria with low levels of intrinsic resistance can, in the absence of relevant selection, become even more susceptible to antibiotics.},
keywords = {Correlated Responses},
pubstate = {published},
tppubtype = {article}
}