2026
Donofrio M A; Blasius H L; Nguyen C C; Schnell A L; Turner C B
Antibiotic susceptibility of Escherichia coli is affected by evolutionary history but not by history of elemental limitation Journal Article
mSphere, vol. 11, iss. 4, pp. e00538-25, 2026.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Descendant Experiments, Genotypes and Phenotypes
@article{Donofrio2026,
title = {Antibiotic susceptibility of \textit{Escherichia coli} is affected by evolutionary history but not by history of elemental limitation},
author = {Marissa A. Donofrio and Heather L. Blasius and Catherine C. Nguyen and Alexa L. Schnell and Caroline B. Turner},
url = {https://journals.asm.org/doi/10.1128/msphere.00538-25},
doi = {10.1128/msphere.00538-25},
year = {2026},
date = {2026-03-23},
urldate = {2026-03-23},
journal = {mSphere},
volume = {11},
issue = {4},
pages = {e00538-25},
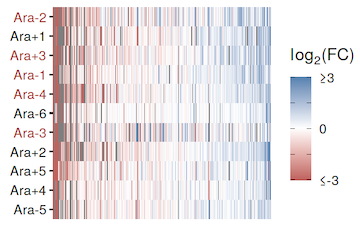
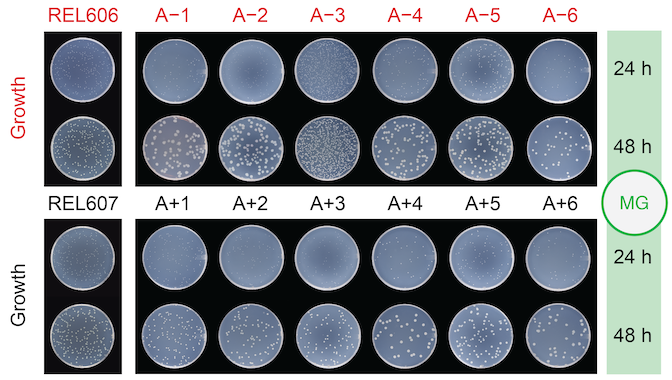
abstract = {ABSTRACT: Antibiotic resistance in bacteria is a global public health threat. To understand how the evolution of antibiotic susceptibility is affected by environmental conditions and prior evolutionary history, we worked with populations from the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}. These populations previously evolved independently for 50,000 generations in an environment without antibiotics, making them an ideal system for studying the effect of evolutionary history on adaptation to new selective pressures. We further evolved five of the LTEE populations, as well as their shared ancestor, under either carbon- or nitrogen-limited conditions and then tested intrinsic resistance to four antibiotics. Evolution under elemental limitation did not have a significant impact on resistance to any of the tested antibiotics. However, some LTEE populations did have higher resistance than other populations. Susceptibility also varied within one population, which had the lowest level of resistance to all four antibiotics. We hypothesized that resistance levels might differ between two clades of bacteria that have coexisted within this population for more than 40,000 generations. Interestingly, although antibiotic susceptibility varied within the population, there was no consistent difference between clades. Instead, one particular clone isolated from the population exhibited higher resistance than the other clones sampled. These findings indicate that antibiotic resistance can vary both within and between experimentally evolved populations, even in the absence of direct selection on resistance. Our results also show that measured levels of susceptibility may depend on stochastic sampling effects during isolation of clones. IMPORTANCE: Antibiotic resistance is one of the most pressing health challenges worldwide, and understanding how bacteria evolve resistance, even when not directly exposed to antibiotics, is critical for managing and predicting emerging threats. Our study leverages the unique Long-Term Evolution Experiment with \textit{Escherichia coli} to show that both the evolutionary history of bacterial populations and random variation among individual clones can significantly influence intrinsic antibiotic susceptibility. Our results also suggest that elemental limitation, while a critical environmental variable, may not be an important driver of intrinsic antibiotic susceptibility, at least over short time frames.},
keywords = {Correlated Responses, Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Ascensao J A; Abedi K D; Prasad A N; Hallatschek O
Frequency-dependent fitness effects are ubiquitous Unpublished
2026.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology
@unpublished{Ascensao2026b,
title = {Frequency-dependent fitness effects are ubiquitous},
author = {Joao A Ascensao and Keon D Abedi and Aditya N Prasad and Oskar Hallatschek},
doi = {10.1101/2025.08.18.670924 },
year = {2026},
date = {2026-02-22},
urldate = {2026-02-22},
abstract = {In simple microbial populations, the fitness effects of selected mutations are typically assumed to be constant, regardless of mutant frequency. This assumption underpins predictions about evolutionary dynamics, epistasis, and the maintenance of genetic diversity. Here, we systematically test this assumption using beneficial mutations from early generations of the \textit{Escherichia coli} Long-Term Evolution Experiment (LTEE). Using flow cytometry-based competition assays, we find that frequency-dependent fitness effects are the norm rather than the exception, occurring in approximately 80% of strain pairs tested. Most competitions exhibit negative frequency-dependence, where fitness advantages decline as mutant frequency increases. We demonstrate that the strength of frequency-dependence is predictable from invasion fitness measurements, which explain approximately half of the biological variation in frequency-dependent slopes. We also observe violations of fitness transitivity in several strain combinations, indicating that competitive outcomes cannot always be predicted from fitness measured against a single reference strain. High-resolution measurements reveal that frequency-dependence changes substantially within the growth cycle, with the net per-cycle effect reflecting the balance of opposing dynamics at different growth phases. Our results demonstrate that even in a model system designed to minimize ecological complexity, subtle interactions between closely related genotypes create frequency-dependent selection that can fundamentally alter evolutionary trajectories.},
keywords = {Demography and Ecology},
pubstate = {published},
tppubtype = {unpublished}
}
Ascensao J A; Yu Q; Hallatschek O
The evolution of genetic drift over 50,000 generations Unpublished
2026.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Theory and Simulations
@unpublished{Ascensao2026a,
title = {The evolution of genetic drift over 50,000 generations},
author = {Joao A. Ascensao and QinQin Yu and Oskar Hallatschek},
doi = {10.64898/2026.01.25.701616 },
year = {2026},
date = {2026-01-27},
urldate = {2026-01-27},
abstract = {Random variation in reproductive success—genetic drift—profoundly shapes genetic diversity and evolutionary trajectories. The strength of drift depends on the variance in descendant number, {$sigma_d^2$}, which governs key evolutionary outcomes: for instance, the establishment probability of a beneficial mutation scales inversely with {$sigma_d^2$} . However, whether {$sigma_d^2$} itself evolves over long timescales has remained unclear, because allele-frequency fluctuations depend on drift only through the effective population size, {$N_e} = N/ {$sigma_d^2$} , which blends census population size with descendant-number variance. Here, we disentangle these components by using model-based Bayesian inference combined with joint tracking of (i) frequency fluctuations of neutrally barcoded lineages and (ii) census population sizes across growth cycles in the E. coli Long-Term Evolution Experiment. Analyzing 33 clones spanning the ancestor through 50,000 generations in two replicate populations (Ara-2 and Ara+2), we find that the strength of genetic drift evolved markedly—and divergently—between the two replicate populations. Both census size and {$sigma_d^2$} changed substantially through time, with most variation in {$N_e} driven by shifts in {$sigma_d^2$} rather than census size. After approximately 2,000 generations, the {$sigma_d^2$}of the two populations diverged sharply: Ara+2 generally remained close to a bottleneck-only null expectation, whereas Ara-2 exhibited 1.5–5×stronger drift, consistent with an evolved increase in stochasticity during growth. Because establishment probability scales as s / {$sigma_d^2$} a beneficial mutation of given effect is roughly twice as likely to establish in Ara+2 as in Ara-2. Our results demonstrate that the key parameter governing genetic drift can itself evolve, with direct consequences for adaptation.},
keywords = {Demography and Ecology, Theory and Simulations},
pubstate = {published},
tppubtype = {unpublished}
}
2025
Lozzi B; Adepoju L; Espinoza J L; Padgen M; Parra M; Ricco A; Castro-Wallace S; Barrick J E; O’Rourke A
Simulated microgravity triggers a membrane adaptation to stress in E. coli REL606 Journal Article
BMC Microbiology, vol. 25, no. 1, pp. 362, 2025, ISSN: 1471-2180.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genotypes and Phenotypes
@article{Lozzi2025,
title = {Simulated microgravity triggers a membrane adaptation to stress in \textit{E. coli} REL606},
author = {Brittney Lozzi and Lea Adepoju and Josh L. Espinoza and Michael Padgen and Macarena Parra and Antonio Ricco and Sarah Castro-Wallace and Jeffrey E. Barrick and Aubrie O’Rourke},
doi = {10.1186/s12866-025-04064-7},
issn = {1471-2180},
year = {2025},
date = {2025-06-09},
urldate = {2025-06-09},
journal = {BMC Microbiology},
volume = {25},
number = {1},
pages = {362},
abstract = {Investigating the evolution of \textit{Escherichia coli} in microgravity offers valuable insights into microbial adaptation to extreme environments. Here the effects of simulated microgravity (SµG) on gene expression and genome evolution of \textit{E. coli} REL606, a strain evolved terrestrially for 35 years, is explored. The transcriptomic changes for glucose-limited and glucose-replete conditions over 24 h illustrate that SµG increased the expression of genes involved in stress response, biofilm, and metabolism. A greater number of differentially expressed genes related to the general stress response (GSR) and biofilm formation is observed in simulated microgravity cultures under glucose-limited conditions in comparison to glucose-replete conditions. Longer term SµG culture under glucose-limited conditions led to the accumulation of unique mutations when compared to control cultures, particularly in the \textit{mraZ}/\textit{fruR} intergenic region and the \textit{elyC} gene, suggesting changes in peptidoglycan and enterobacterial common antigen (ECA) production. These findings highlight the physiological and genomic adaptations of E. coli to microgravity, offering a foundation for future research into the long-term effects of space conditions on bacterial evolution.},
keywords = {Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Chihoub D; Pintard C; Lenski R E; Tenaillon O; Couce A
The evolution of robustness and fragility during long-term bacterial adaptation Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 122, no. 16, pp. e2501901122, 2025.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Fitness Trajectories
@article{Chihoub2025,
title = {The evolution of robustness and fragility during long-term bacterial adaptation},
author = {Doha Chihoub and Coralie Pintard and Richard E. Lenski and Olivier Tenaillon and Alejandro Couce},
doi = {10.1073/pnas.2501901122},
year = {2025},
date = {2025-01-27},
urldate = {2025-01-27},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {122},
number = {16},
pages = {e2501901122},
abstract = {Theory predicts that well-adapted populations may evolve mechanisms to counteract the inevitable influx of deleterious mutations. While mutational robustness can be directly selected in the laboratory, evidence for its spontaneous evolution during general adaptation is mixed. Moreover, whether robustness evolves to include pleiotropic effects remains largely unexplored. Here, we studied the effects of point mutations in the RNA polymerase of \textit{Escherichia coli} over a 15,000-generation adaptive trajectory. Fitness effects of both beneficial and deleterious mutations were attenuated in fitter backgrounds. In contrast, pleiotropic effects became more severe and widespread with greater adaptation. These results show that trade-offs between robustness and fragility can evolve in regulatory networks, regardless of whether driven by adaptive or nonadaptive processes. More broadly, they illustrate how adaptation can generate hidden variability, with unpredictable evolutionary consequences in new environments.},
keywords = {Correlated Responses, Fitness Trajectories},
pubstate = {published},
tppubtype = {article}
}
2024
Izutsu M; Lake D M; Matson Z W D; Dodson J P; Lenski R E
Effects of periodic bottlenecks on the dynamics of adaptive evolution in microbial populations Journal Article
Microbiology (Reading, England), vol. 170, no. 9, pp. 001494, 2024, ISSN: 1465-2080.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Theory and Simulations
@article{Izutsu_2024,
title = {Effects of periodic bottlenecks on the dynamics of adaptive evolution in microbial populations},
author = {Minako Izutsu and Devin M. Lake and Zachary W. D. Matson and Jack P. Dodson and Richard E. Lenski},
doi = {10.1099/mic.0.001494},
issn = {1465-2080},
year = {2024},
date = {2024-09-18},
urldate = {2021-12-30},
journal = {Microbiology (Reading, England)},
volume = {170},
number = {9},
pages = {001494},
publisher = {LTEE},
abstract = {Population bottlenecks can impact the rate of adaptation in evolving populations. On the one hand, each bottleneck reduces the genetic variation that fuels adaptation. On the other hand, each founder that survives a bottleneck can undergo more generations and leave more descendants in a resource-limited environment, which allows surviving beneficial mutations to spread more quickly. A theoretical model predicted that the rate of fitness gains should be maximized using ~8-fold dilutions. Here we investigate the impact of repeated bottlenecks on the dynamics of adaptation using numerical simulations and experimental populations of Escherichia coli. Our simulations confirm the model's prediction when populations evolve in a regime where beneficial mutations are rare and waiting times between successful mutations are long. However, more extreme dilutions maximize fitness gains in simulations when beneficial mutations are common and clonal interference prevents most of them from fixing. To examine these predictions, we propagated 48 \textit{E. coli} populations with 2-, 8-, 100-, and 1000-fold dilutions for 150 days. Adaptation began earlier and fitness gains were greater with 100- and 1000-fold dilutions than with 8-fold dilutions, consistent with the simulations when beneficial mutations are common. However, the selection pressures in the 2-fold treatment were qualitatively different from the other treatments, violating a critical assumption of the model and simulations. Thus, varying the dilution factor during periodic bottlenecks can have multiple effects on the dynamics of adaptation caused by differential losses of diversity, different numbers of generations, and altered selection.},
keywords = {Descendant Experiments, Fitness Trajectories, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}
uz-Zaman Md H; D'Alton S; Barrick J E; Ochman H
Promoter capture drives the emergence of proto-genes in Escherichia coli Journal Article
PLoS Biology, vol. 22, pp. e3002418, 2024.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution
@article{uz-Zaman2024,
title = {Promoter capture drives the emergence of proto-genes in \textit{Escherichia coli}},
author = {Md. Hassan uz-Zaman and Simon D'Alton and Jeffrey E. Barrick and Howard Ochman},
doi = {10.1371/journal.pbio.3002418},
year = {2024},
date = {2024-05-07},
urldate = {2023-11-17},
journal = {PLoS Biology},
volume = {22},
pages = {e3002418},
publisher = {Cold Spring Harbor Laboratory},
abstract = {The phenomenon of de novo gene birth-the emergence of genes from non-genic sequences-has received considerable attention due to the widespread occurrence of genes that are unique to particular species or genomes. Most instances of de novo gene birth have been recognized through comparative analyses of genome sequences in eukaryotes, despite the abundance of novel, lineage-specific genes in bacteria and the relative ease with which bacteria can be studied in an experimental context. Here, we explore the genetic record of the \textit{Escherichia coli} long-term evolution experiment (LTEE) for changes indicative of "proto-genic" phases of new gene birth in which non-genic sequences evolve stable transcription and/or translation. Over the time span of the LTEE, non-genic regions are frequently transcribed, translated and differentially expressed, with levels of transcription across low-expressed regions increasing in later generations of the experiment. Proto-genes formed downstream of new mutations result either from insertion element activity or chromosomal translocations that fused preexisting regulatory sequences to regions that were not expressed in the LTEE ancestor. Additionally, we identified instances of proto-gene emergence in which a previously unexpressed sequence was transcribed after formation of an upstream promoter, although such cases were rare compared to those caused by recruitment of preexisting promoters. Tracing the origin of the causative mutations, we discovered that most occurred early in the history of the LTEE, often within the first 20,000 generations, and became fixed soon after emergence. Our findings show that proto-genes emerge frequently within evolving populations, can persist stably, and can serve as potential substrates for new gene formation. },
keywords = {Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
Mori M; Patsalo V; Euler C; Williamson J R; Scott M
Proteome partitioning constraints in long-term laboratory evolution Journal Article
Nature Communications, vol. 15, pp. 4087, 2024, ISSN: 2041-1723.
Abstract | Links | BibTeX | Altmetric | Tags: experimental evolution, Genotypes and Phenotypes, Microbial genetics, Proteomic analysis
@article{Mori2024,
title = {Proteome partitioning constraints in long-term laboratory evolution},
author = {Matteo Mori and Vadim Patsalo and Christian Euler and James R. Williamson and Matthew Scott},
url = {https://www.nature.com/articles/s41467-024-48447-2},
doi = {10.1038/s41467-024-48447-2},
issn = {2041-1723},
year = {2024},
date = {2024-05-01},
urldate = {2024-05-01},
journal = {Nature Communications},
volume = {15},
pages = {4087},
abstract = {Adaptive laboratory evolution experiments provide a controlled context in which the dynamics of selection and adaptation can be followed in real-time at the single-nucleotide level. And yet this precision introduces hundreds of degrees-of-freedom as genetic changes accrue in parallel lineages over generations. On short timescales, physiological constraints have been leveraged to provide a coarse-grained view of bacterial gene expression characterized by a small set of phenomenological parameters. Here, we ask whether this same framework, operating at a level between genotype and fitness, informs physiological changes that occur on evolutionary timescales. Using a strain adapted to growth in glucose minimal medium, we find that the proteome is substantially remodeled over 40 000 generations. The most striking change is an apparent increase in enzyme efficiency, particularly in the enzymes of lower-glycolysis. We propose that deletion of metabolic flux-sensing regulation early in the adaptation results in increased enzyme saturation and can account for the observed proteome remodeling.},
keywords = {experimental evolution, Genotypes and Phenotypes, Microbial genetics, Proteomic analysis},
pubstate = {published},
tppubtype = {article}
}
Staples R K; Cooper T F
2024.
Abstract | Links | BibTeX | Altmetric | Tags:
@unpublished{Staples2024,
title = {Mutations in \textit{iclR} increase evolvability by facilitating compensation that exposes cryptic beneficial mutations in experimental populations of \textit{Escherichia coli }},
author = { Rachel K. Staples AND Tim F. Cooper},
doi = {10.1101/2024.02.21.581449},
year = {2024},
date = {2024-02-21},
urldate = {2024-02-21},
abstract = {Evolvability describes the potential of a population to generate beneficial variation. Several mechanisms that increase evolvability have been demonstrated, including the action of systems that reveal accumulated beneficial variants following an environmental shift. We examine the basis of an increase in the evolvability of \textit{Escherichia coli} lines that were first selected in an environment supplemented with glucose as sole carbon source and then transferred to an otherwise identical lactose supplemented environment. These lines increased in fitness significantly more quickly in the lactose environment, and reached a higher final fitness, than did naïve ancestral lines. In four of six lines this increased evolvability can be explained by mutations in \textit{iclR} that were selected in glucose but were significantly deleterious in lactose, masking the effect of other generally beneficial mutations. Secondary mutations that compensated for this cost resulted in large fitness increases. We did not detect any consistent genetic signature associated with the compensation, suggesting that different pathways were responsible and, therefore, that it can occur at a relatively high rate. That mutations selected in one environment will become deleterious following an environmental shift, so that compensation provides potential for a large subsequent fitness increase represents a potentially common and general mechanism of evolvability in changing environments.
},
keywords = {},
pubstate = {published},
tppubtype = {unpublished}
}
Ascensao J A; Denk J; Lok K; Yu Q; Wetmore K M; Hallatschek O
Rediversification following ecotype isolation reveals hidden adaptive potential Journal Article
Current Biology, vol. 34, no. 4, pp. 855–867, 2024, ISSN: 0960-9822.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Genotypes and Phenotypes, Historical Contingency
@article{Ascensao2024,
title = {Rediversification following ecotype isolation reveals hidden adaptive potential},
author = {Joao A. Ascensao and Jonas Denk and Kristen Lok and QinQin Yu and Kelly M. Wetmore and Oskar Hallatschek},
url = {https://www.sciencedirect.com/science/article/pii/S0960982224000290},
doi = {10.1016/j.cub.2024.01.029},
issn = {0960-9822},
year = {2024},
date = {2024-02-01},
urldate = {2024-02-01},
journal = {Current Biology},
volume = {34},
number = {4},
pages = {855--867},
abstract = {Microbial communities play a critical role in ecological processes, and their diversity is key to their functioning. However, little is known about whether communities can regenerate ecological diversity following ecotype removal or extinction and how the rediversified communities would compare to the original ones. Here, we show that simple two-ecotype communities from the \textit{E. coli} long-term evolution experiment (LTEE) consistently rediversified into two ecotypes following the isolation of one of the ecotypes, coexisting via negative frequency-dependent selection. Communities separated by more than 30,000 generations of evolutionary time rediversify in similar ways. The rediversified ecotype appears to share a number of growth traits with the ecotype it replaces. However, the rediversified community is also different from the original community in ways relevant to the mechanism of ecotype coexistence—for example, in stationary phase response and survival. We found substantial variation in the transcriptional states between the two original ecotypes, whereas the differences within the rediversified community were comparatively smaller, although the rediversified community showed unique patterns of differential expression. Our results suggest that evolution may leave room for alternative diversification processes even in a maximally reduced community of only two strains. We hypothesize that the presence of alternative evolutionary pathways may be even more pronounced in communities of many species where there are even more potential niches, highlighting an important role for perturbations, such as species removal, in evolving ecological communities.},
keywords = {Demography and Ecology, Genotypes and Phenotypes, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Couce A; Limdi A; Magnan M; Owen S V; Herren C M; Lenski R E; Tenaillon O; Baym M
Changing fitness effects of mutations through long-term bacterial evolution Journal Article
Science, vol. 383, no. 6681, pp. eadd1417, 2024.
Abstract | Links | BibTeX | Altmetric | Tags:
@article{Couce2024,
title = {Changing fitness effects of mutations through long-term bacterial evolution},
author = {Alejandro Couce and Anurag Limdi and Melanie Magnan and Siân V. Owen and Cristina M. Herren and Richard E. Lenski and Olivier Tenaillon and Michael Baym},
url = {https://www.science.org/doi/10.1126/science.add1417},
doi = {10.1126/science.add1417},
year = {2024},
date = {2024-01-26},
urldate = {2024-01-26},
journal = {Science},
volume = {383},
number = {6681},
pages = {eadd1417},
abstract = {The distribution of fitness effects of new mutations shapes evolution, but it is challenging to observe how it changes as organisms adapt. Using \textit{Escherichia coli} lineages spanning 50,000 generations of evolution, we quantify the fitness effects of insertion mutations in every gene. Macroscopically, the fraction of deleterious mutations changed little over time whereas the beneficial tail declined sharply, approaching an exponential distribution. Microscopically, changes in individual gene essentiality and deleterious effects often occurred in parallel; altered essentiality is only partly explained by structural variation. The identity and effect sizes of beneficial mutations changed rapidly over time, but many targets of selection remained predictable because of the importance of loss-of-function mutations. Taken together, these results reveal the dynamic—but statistically predictable—nature of mutational fitness effects.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2023
Favate J S; Skalenko K S; Chiles E; Su X; Yadavalli S S; Shah P
Linking genotypic and phenotypic changes in the E. coli long-term evolution experiment using metabolomics Journal Article
eLife, vol. 12, pp. RP87039, 2023, ISSN: eLife.
Abstract | Links | BibTeX | Altmetric | Tags: Genotypes and Phenotypes, Parallelism and Divergence
@article{10.7554/eLife.87039,
title = {Linking genotypic and phenotypic changes in the \textit{E. coli} long-term evolution experiment using metabolomics},
author = {John S Favate and Kyle S Skalenko and Eric Chiles and Xiaoyang Su and Srujana Samhita Yadavalli and Premal Shah},
editor = {John McCutcheon and Christian R Landry},
url = {https://doi.org/10.7554/eLife.87039},
doi = {10.7554/eLife.87039},
issn = {eLife},
year = {2023},
date = {2023-11-01},
urldate = {2023-11-01},
journal = {eLife},
volume = {12},
pages = {RP87039},
publisher = {2050-084X},
abstract = {Changes in an organism's environment, genome, or gene expression patterns can lead to changes in its metabolism. The metabolic phenotype can be under selection and contributes to adaptation. However, the networked and convoluted nature of an organism's metabolism makes relating mutations, metabolic changes, and effects on fitness challenging. To overcome this challenge, we use the long-term evolution experiment (LTEE) with \textit{E. coli} as a model to understand how mutations can eventually affect metabolism and perhaps fitness. We used mass spectrometry to broadly survey the metabolomes of the ancestral strains and all 12 evolved lines. We combined this metabolic data with mutation and expression data to suggest how mutations that alter specific reaction pathways, such as the biosynthesis of nicotinamide adenine dinucleotide, might increase fitness in the system. Our work provides a better understanding of how mutations might affect fitness through the metabolic changes in the LTEE and thus provides a major step in developing a complete genotype-phenotype map for this experimental system. },
key = {E. coli; LTEE; adaptation; bacteria; evolutionary biology; metabolomics},
keywords = {Genotypes and Phenotypes, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
Barrick J E; Blount Z D; Lake D M; Dwenger J H; Chavarria-Palma J E; Izutsu M; Wiser M J
Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Journal of Visualized Experiments, vol. 198, pp. e65342, 2023, ISSN: 1940-087X.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Methods and Miscellaneous
@article{Barrick2023,
title = {Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli},
author = {Jeffrey E. Barrick and Zachary D. Blount and Devin M. Lake and Jack H. Dwenger and Jesus E. Chavarria-Palma and Minako Izutsu and Michael J. Wiser},
doi = {10.3791/65342},
issn = {1940-087X},
year = {2023},
date = {2023-08-18},
urldate = {2023-08-18},
journal = {Journal of Visualized Experiments},
volume = {198},
pages = {e65342},
abstract = {The Long-Term Evolution Experiment (LTEE) has followed twelve populations of \textit{Escherichia coli} as they have adapted to a simple laboratory environment for more than 35 years and 77,000 bacterial generations. The setup and procedures used in the LTEE epitomize reliable and reproducible methods for studying microbial evolution. In this protocol, we first describe how the LTEE populations are transferred to fresh medium and cultured each day. Then, we describe how the LTEE populations are regularly checked for possible signs of contamination and archived to provide a permanent frozen "fossil record" for later study. Multiple safeguards included in these procedures are designed to prevent contamination, detect various problems when they occur, and recover from disruptions without appreciably setting back the progress of the experiment. One way that the overall tempo and character of evolutionary changes are monitored in the LTEE is by measuring the competitive fitness of populations and strains from the experiment. We describe how co-culture competition assays are conducted and provide both a spreadsheet and an R package (fitnessR) for calculating relative fitness from the results. Over the course of the LTEE, the behaviors of some populations have changed in interesting ways, and new technologies like whole-genome sequencing have provided additional avenues for investigating how the populations have evolved. We end by discussing how the original LTEE procedures have been updated to accommodate or take advantage of these changes. This protocol will be useful for researchers who use the LTEE as a model system for studying connections between evolution and genetics, molecular biology, systems biology, and ecology. More broadly, the LTEE provides a tried-and-true template for those who are beginning their own evolution experiments with new microbes, environments, and questions. },
keywords = {Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
Turner C B; Blount Z D; Mitchell D H; Lenski R E
Evolution of a cross-feeding interaction following a key innovation in a long-term evolution experiment with Escherichia coli Journal Article
Microbiology (Reading), vol. 169, no. 8, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Demography and Ecology, Historical Contingency
@article{Turner2023,
title = { Evolution of a cross-feeding interaction following a key innovation in a long-term evolution experiment with \textit{Escherichia coli} },
author = {Caroline B. Turner and Zachary D. Blount and Daniel H. Mitchell and Richard E. Lenski},
doi = {10.1099/mic.0.001390},
year = {2023},
date = {2023-08-01},
urldate = {2023-08-01},
journal = { Microbiology (Reading)},
volume = {169},
number = {8},
abstract = {The evolution of a novel trait can profoundly change an organism's effects on its environment, which can in turn affect the further evolution of that organism and any coexisting organisms. We examine these effects and feedbacks following the evolution of a novel function in the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}. A characteristic feature of \textit{E. coli} is its inability to grow aerobically on citrate (Cit^{−}). Nonetheless, a Cit^{+} variant with this capacity evolved in one LTEE population after 31 000 generations. The Cit^{+} clade then coexisted stably with another clade that retained the ancestral Cit^{−} phenotype. This coexistence was shaped by the evolution of a cross-feeding relationship based on C4-dicarboxylic acids, particularly succinate, fumarate, and malate, that the Cit^{+} variants release into the medium. Both the Cit^{−} and Cit^{+} cells evolved to grow on these excreted resources. The evolution of aerobic growth on citrate thus led to a transition from an ecosystem based on a single limiting resource, glucose, to one with at least five resources that were either shared or partitioned between the two coexisting clades. Our findings show that evolutionary novelties can change environmental conditions in ways that facilitate diversity by altering ecosystem structure and the evolutionary trajectories of coexisting lineages. },
keywords = {Citrate Evolution, Demography and Ecology, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Mukherjee A; Ealy J; Huang Y; Benites N C; Polk M; Basan M
Coexisting ecotypes in long-term evolution emerged from interacting trade-offs Journal Article
Nature Communications, vol. 14, no. 1, pp. 3805, 2023, ISSN: 2041-1723.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Theory and Simulations
@article{Mukherjee2023,
title = {Coexisting ecotypes in long-term evolution emerged from interacting trade-offs},
author = {Avik Mukherjee and Jade Ealy and Yanqing Huang and Nina Catherine Benites and Mark Polk and Markus Basan},
url = {https://www.nature.com/articles/s41467-023-39471-9},
doi = {10.1038/s41467-023-39471-9},
issn = {2041-1723},
year = {2023},
date = {2023-06-26},
journal = {Nature Communications},
volume = {14},
number = {1},
pages = {3805},
abstract = {Evolution of complex communities of coexisting microbes remains poorly understood. The long-term evolution experiment on \textit{Escherichia coli} (LTEE) revealed the spontaneous emergence of stable coexistence of multiple ecotypes, which persisted for more than 14,000 generations of continuous evolution. Here, using a combination of experiments and computer simulations, we show that the emergence and persistence of this phenomenon can be explained by the combination of two interacting trade-offs, rooted in biochemical constraints: First, faster growth is enabled by higher fermentation and obligate acetate excretion. Second, faster growth results in longer lag times when utilizing acetate after glucose is depleted. This combination creates an ecological niche for a slower-growing ecotype, specialized in switching to acetate. These findings demonstrate that trade-offs can give rise to surprisingly complex communities with evolutionarily stable coexistence of multiple variants in even the simplest environments.},
keywords = {Demography and Ecology, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}
Lenski R E
Revisiting the design of the long-term evolution experiment with Escherichia coli Journal Article
Journal of Molecular Evolution, vol. 91, no. 3, pp. 241-253, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles
@article{lenski2023,
title = {Revisiting the design of the long-term evolution experiment with \textit{Escherichia coli}},
author = {Richard E. Lenski},
url = {https://link.springer.com/epdf/10.1007/s00239-023-10095-3?sharing_token=zmDHuK0kbvnJBQq1k96fe_e4RwlQNchNByi7wbcMAY53KNkhv6F2YgRIeC8sZGNejxJrvlAGZWInruED5Dqdai5WeU2RAWL2PJNp0pL9QJO39B_ijCtRZcaW8jqM7PclDJfFwL_78U5zNlQYyCOsQwa1Yxha61uXUWhW-Buiq7o=},
doi = {10.1007/s00239-023-10095-3},
year = {2023},
date = {2023-06-01},
urldate = {2023-02-15},
journal = {Journal of Molecular Evolution},
volume = {91},
number = {3},
pages = {241-253},
abstract = {The long-term evolution experiment (LTEE) with \textit{Escherichia coli} began in 1988 and it continues to this day, with its 12 populations having recently reached 75,000 generations of evolution in a simple, well-controlled environment. The LTEE was designed to explore open-ended questions about the dynamics and repeatability of phenotypic and genetic evolution. Here I discuss various aspects of the LTEE’s experimental design that have enabled its stability and success, including the choices of the culture regime, growth medium, ancestral strain, and statistical replication. I also discuss some of the challenges associated with a long-running project, such as handling procedural errors (e.g., cross-contamination) and managing the expanding collection of frozen samples. The simplicity of the experimental design and procedures have supported the long-term stability of the LTEE. That stability—along with the inherent creativity of the evolutionary process and the emergence of new genomic technologies—provides a platform that has allowed talented students and collaborators to pose questions, collect data, and make discoveries that go far beyond anything I could have imagined at the start of the LTEE.},
keywords = {Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles},
pubstate = {published},
tppubtype = {article}
}
Jagdish T
The dynamics of fitness and pleiotropy in a long-term evolution experiment with Escherichia coli PhD Thesis
2023.
Abstract | Links | BibTeX | Tags: Correlated Responses, Demography and Ecology, Fitness Trajectories, Methods and Miscellaneous
@phdthesis{nokey,
title = {The dynamics of fitness and pleiotropy in a long-term evolution experiment with \textit{Escherichia coli}},
author = {Tanush Jagdish},
url = {https://dash.harvard.edu/handle/1/37375541},
year = {2023},
date = {2023-05-08},
urldate = {2023-05-08},
abstract = {Life on earth is shaped by a delicate balance between chance and necessity. A combination of natural selection and genetic drift has moulded random genetic variations over the course of billions of years to generate the complex and interconnected biosphere we see today. These evolutionary dynamics are ultimately the product of numerous genetic mechanisms operating in genomes with highly complex architectures. Unravelling the rules of genetic mechanisms responsible for evolutionary innovation is critical to developing a comprehensive and predictive evolutionary theory, but has evaded direct experimentation since the scale of resources and technology needed to study the patterns of genetic evolution has only recently become achievable. In this dissertation, I explore the use of microbial experimental evolution as a powerful tool for probing evolutionary questions, particularly by leveraging DNA-barcoding and high-throughput DNA sequencing. In chapter 1, I provide an overview of the field of microbial experimental evolution, dividing its history into two eras marked by qualitatively different methodological advancements. I make the case that we now stand at the dawn of a third era, where advances in genome-engineering coupled with low-cost, high-throughput DNA sequencing will allow experiments to finally probe evolution on a statistical scale. I then present two research studies that take advantage of microbial experimental evolution to investigate distinct genetic mechanisms key to the evolutionary process. In Chapter 2, I explore whether fitness can continue increasing in a population that has already adapted to its environment for over 30,000 generations, and whether fixations of beneficial mutations from population-wide DNA sequencing can predict jumps in fitness. My findings reveal that fitness continues to monotonically increase in step with the fixation of beneficial mutations, even though the rate of fixation has dramatically slowed down, highlighting the potential for ongoing adaptation even under constant environmental conditions. In Chapter 3, I develop a novel conjugation-based DNA-barcoding method for the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}, allowing me to examine the pleiotropic consequences of adaptation to glucose over 50,000 generations in 15 novel resource environments. My observations reveal broad patterns of both convergent and divergent evolution that correspond with mutations in key metabolic genes in clonal sequencing datasets, shedding light on the nature of pleiotropy and its evolution over extended timescales. Using microbial model systems in an evolutionary context has the unique advantage of being relevant both to fundamental evolutionary biology and human health. Since genetic drift and rare mutational events both play an outsized role in determining the evolutionary trajectories of populations, evolutionary questions in the modern age will increasingly be faced with issues of scale. Microbial experimental evolution offers both scale and tractability to solve this problem. Uniquely, this does not sacrifice on human relevance. Building a coarse-grained and comprehensive evolutionary theory is more significant to society today than ever before as the importance of clonal evolution in cancer, gut microbiomes and even pandemics becomes more clearly understood.},
keywords = {Correlated Responses, Demography and Ecology, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {phdthesis}
}
Ascensao J A; Wetmore K M; Good B H; Arkin A P; Hallatschek O
Quantifying the adaptive potential of a nascent bacterial community Journal Article
Nature Communications, vol. 14, no. 1, pp. 248, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Descendant Experiments, Genome Evolution, Genotypes and Phenotypes
@article{ascensao2023,
title = {Quantifying the adaptive potential of a nascent bacterial community},
author = {Joao A. Ascensao and Kelly M. Wetmore and Benjamin H. Good and Adam P. Arkin and Oskar Hallatschek},
url = {https://www.nature.com/articles/s41467-022-35677-5},
doi = {10.1038/s41467-022-35677-5},
year = {2023},
date = {2023-01-16},
urldate = {2022-01-01},
journal = {Nature Communications},
volume = {14},
number = {1},
pages = {248},
publisher = {Cold Spring Harbor Laboratory},
abstract = {The fitness effects of all possible mutations available to an organism largely shape the dynamics of evolutionary adaptation. Yet, whether and how this adaptive landscape changes over evolutionary times, especially upon ecological diversification and changes in community composition, remains poorly understood. We sought to fill this gap by analyzing a stable community of two closely related ecotypes (“L” and “S”) shortly after they emerged within the \textit{E. coli} Long-Term Evolution Experiment (LTEE). We engineered genome-wide barcoded transposon libraries to measure the invasion fitness effects of all possible gene knockouts in the coexisting strains as well as their ancestor, for many different, ecologically relevant conditions. We find consistent statistical patterns of fitness effect variation across both genetic background and community composition, despite the idiosyncratic behavior of individual knockouts. Additionally, fitness effects are correlated with evolutionary outcomes for a number of conditions, possibly revealing shifting patterns of adaptation. Together, our results reveal how ecological and epistatic effects combine to shape the adaptive landscape in a nascent ecological community.},
keywords = {Demography and Ecology, Descendant Experiments, Genome Evolution, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2022
Laurin D; Mercier C; Quansah N; Robert J S; Usson Y; Schneider D; Hindré T; Schaack B
Extracellular vesicles from 50,000 generation clones of the Escherichia coli long-term evolution experiment Journal Article
International Journal of Molecular Sciences, vol. 23, pp. 14580, 2022, ISSN: 1422-0067.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Genotypes and Phenotypes
@article{Laurin2022,
title = {Extracellular vesicles from 50,000 generation clones of the \textit{Escherichia coli} long-term evolution experiment},
author = {David Laurin and Corinne Mercier and Nyamekye Quansah and Julie Suzanne Robert and Yves Usson and Dominique Schneider and Thomas Hindré and Béatrice Schaack },
url = {https://www.mdpi.com/1422-0067/23/23/14580},
doi = {10.3390/ijms232314580},
issn = {1422-0067},
year = {2022},
date = {2022-11-01},
urldate = {2022-11-01},
journal = {International Journal of Molecular Sciences},
volume = {23},
pages = {14580},
abstract = {Extracellular vesicles (EVs) are critical elements of cell–cell communication. Here, we characterized the outer membrane vesicles (OMVs) released by specific clones of Escherichia coli isolated from the Long-Term Evolution Experiment after 50,000 generations (50K) of adaptation to glucose minimal medium. Compared with their ancestor, the evolved clones produce small OMVs but also larger ones which display variable amounts of both OmpA and LPS. Tracking ancestral, fluorescently labelled OMVs revealed that they fuse with both ancestral- and 50K-evolved cells, albeit in different proportions. We quantified that less than 2% of the cells from one 50K-evolved clone acquired the fluorescence delivered by OMVs from the ancestral strain but that one cell concomitantly fuses with several OMVs. Globally, our results showed that OMV production in E. coli is a phenotype that varies along bacterial evolution and question the contribution of OMVs-mediated interactions in bacterial adaptation.},
keywords = {Cell Morphology, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Favate J S; Liang S; Cope A L; Yadavilli S S; Shah P
The landscape of transcriptional and translational changes over 22 years of bacterial adaptation Journal Article
eLife, vol. 11, pp. e81979, 2022.
Abstract | Links | BibTeX | Altmetric | Tags: Genotypes and Phenotypes, Parallelism and Divergence
@article{favate2022,
title = {The landscape of transcriptional and translational changes over 22 years of bacterial adaptation},
author = {John S Favate and Shun Liang and Alexander L Cope and Srujana S Yadavilli and Premal Shah},
editor = {Detlef Weigel},
url = {https://elifesciences.org/articles/81979},
doi = {10.7554/eLife.81979},
year = {2022},
date = {2022-10-10},
urldate = {2022-10-10},
journal = {eLife},
volume = {11},
pages = {e81979},
abstract = {Organisms can adapt to an environment by taking multiple mutational paths. This redundancy at the genetic level, where many mutations have similar phenotypic and fitness effects, can make untangling the molecular mechanisms of complex adaptations difficult. Here, we use the \textit{Escherichia coli} long-term evolution experiment (LTEE) as a model to address this challenge. To understand how different genomic changes could lead to parallel fitness gains, we characterize the landscape of transcriptional and translational changes across 12 replicate populations evolving in parallel for 50,000 generations. By quantifying absolute changes in mRNA abundances, we show that not only do all evolved lines have more mRNAs but that this increase in mRNA abundance scales with cell size. We also find that despite few shared mutations at the genetic level, clones from replicate populations in the LTEE are remarkably similar in their gene expression patterns at both the transcriptional and translational levels. Furthermore, we show that the majority of the expression changes are due to changes at the transcriptional level with very few translational changes. Finally, we show how mutations in transcriptional regulators lead to consistent and parallel changes in the expression levels of downstream genes. These results deepen our understanding of the molecular mechanisms underlying complex adaptations and provide insights into the repeatability of evolution.},
howpublished = {eLife},
keywords = {Genotypes and Phenotypes, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}