2026
Donofrio M A; Blasius H L; Nguyen C C; Schnell A L; Turner C B
Antibiotic susceptibility of Escherichia coli is affected by evolutionary history but not by history of elemental limitation Journal Article
mSphere, vol. 11, iss. 4, pp. e00538-25, 2026.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Descendant Experiments, Genotypes and Phenotypes
@article{Donofrio2026,
title = {Antibiotic susceptibility of \textit{Escherichia coli} is affected by evolutionary history but not by history of elemental limitation},
author = {Marissa A. Donofrio and Heather L. Blasius and Catherine C. Nguyen and Alexa L. Schnell and Caroline B. Turner},
url = {https://journals.asm.org/doi/10.1128/msphere.00538-25},
doi = {10.1128/msphere.00538-25},
year = {2026},
date = {2026-03-23},
urldate = {2026-03-23},
journal = {mSphere},
volume = {11},
issue = {4},
pages = {e00538-25},
abstract = {ABSTRACT: Antibiotic resistance in bacteria is a global public health threat. To understand how the evolution of antibiotic susceptibility is affected by environmental conditions and prior evolutionary history, we worked with populations from the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}. These populations previously evolved independently for 50,000 generations in an environment without antibiotics, making them an ideal system for studying the effect of evolutionary history on adaptation to new selective pressures. We further evolved five of the LTEE populations, as well as their shared ancestor, under either carbon- or nitrogen-limited conditions and then tested intrinsic resistance to four antibiotics. Evolution under elemental limitation did not have a significant impact on resistance to any of the tested antibiotics. However, some LTEE populations did have higher resistance than other populations. Susceptibility also varied within one population, which had the lowest level of resistance to all four antibiotics. We hypothesized that resistance levels might differ between two clades of bacteria that have coexisted within this population for more than 40,000 generations. Interestingly, although antibiotic susceptibility varied within the population, there was no consistent difference between clades. Instead, one particular clone isolated from the population exhibited higher resistance than the other clones sampled. These findings indicate that antibiotic resistance can vary both within and between experimentally evolved populations, even in the absence of direct selection on resistance. Our results also show that measured levels of susceptibility may depend on stochastic sampling effects during isolation of clones. IMPORTANCE: Antibiotic resistance is one of the most pressing health challenges worldwide, and understanding how bacteria evolve resistance, even when not directly exposed to antibiotics, is critical for managing and predicting emerging threats. Our study leverages the unique Long-Term Evolution Experiment with \textit{Escherichia coli} to show that both the evolutionary history of bacterial populations and random variation among individual clones can significantly influence intrinsic antibiotic susceptibility. Our results also suggest that elemental limitation, while a critical environmental variable, may not be an important driver of intrinsic antibiotic susceptibility, at least over short time frames.},
keywords = {Correlated Responses, Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2025
Lozzi B; Adepoju L; Espinoza J L; Padgen M; Parra M; Ricco A; Castro-Wallace S; Barrick J E; O’Rourke A
Simulated microgravity triggers a membrane adaptation to stress in E. coli REL606 Journal Article
BMC Microbiology, vol. 25, no. 1, pp. 362, 2025, ISSN: 1471-2180.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genotypes and Phenotypes
@article{Lozzi2025,
title = {Simulated microgravity triggers a membrane adaptation to stress in \textit{E. coli} REL606},
author = {Brittney Lozzi and Lea Adepoju and Josh L. Espinoza and Michael Padgen and Macarena Parra and Antonio Ricco and Sarah Castro-Wallace and Jeffrey E. Barrick and Aubrie O’Rourke},
doi = {10.1186/s12866-025-04064-7},
issn = {1471-2180},
year = {2025},
date = {2025-06-09},
urldate = {2025-06-09},
journal = {BMC Microbiology},
volume = {25},
number = {1},
pages = {362},
abstract = {Investigating the evolution of \textit{Escherichia coli} in microgravity offers valuable insights into microbial adaptation to extreme environments. Here the effects of simulated microgravity (SµG) on gene expression and genome evolution of \textit{E. coli} REL606, a strain evolved terrestrially for 35 years, is explored. The transcriptomic changes for glucose-limited and glucose-replete conditions over 24 h illustrate that SµG increased the expression of genes involved in stress response, biofilm, and metabolism. A greater number of differentially expressed genes related to the general stress response (GSR) and biofilm formation is observed in simulated microgravity cultures under glucose-limited conditions in comparison to glucose-replete conditions. Longer term SµG culture under glucose-limited conditions led to the accumulation of unique mutations when compared to control cultures, particularly in the \textit{mraZ}/\textit{fruR} intergenic region and the \textit{elyC} gene, suggesting changes in peptidoglycan and enterobacterial common antigen (ECA) production. These findings highlight the physiological and genomic adaptations of E. coli to microgravity, offering a foundation for future research into the long-term effects of space conditions on bacterial evolution.},
keywords = {Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2024
Izutsu M; Lake D M; Matson Z W D; Dodson J P; Lenski R E
Effects of periodic bottlenecks on the dynamics of adaptive evolution in microbial populations Journal Article
Microbiology (Reading, England), vol. 170, no. 9, pp. 001494, 2024, ISSN: 1465-2080.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Theory and Simulations
@article{Izutsu_2024,
title = {Effects of periodic bottlenecks on the dynamics of adaptive evolution in microbial populations},
author = {Minako Izutsu and Devin M. Lake and Zachary W. D. Matson and Jack P. Dodson and Richard E. Lenski},
doi = {10.1099/mic.0.001494},
issn = {1465-2080},
year = {2024},
date = {2024-09-18},
urldate = {2021-12-30},
journal = {Microbiology (Reading, England)},
volume = {170},
number = {9},
pages = {001494},
publisher = {LTEE},
abstract = {Population bottlenecks can impact the rate of adaptation in evolving populations. On the one hand, each bottleneck reduces the genetic variation that fuels adaptation. On the other hand, each founder that survives a bottleneck can undergo more generations and leave more descendants in a resource-limited environment, which allows surviving beneficial mutations to spread more quickly. A theoretical model predicted that the rate of fitness gains should be maximized using ~8-fold dilutions. Here we investigate the impact of repeated bottlenecks on the dynamics of adaptation using numerical simulations and experimental populations of Escherichia coli. Our simulations confirm the model's prediction when populations evolve in a regime where beneficial mutations are rare and waiting times between successful mutations are long. However, more extreme dilutions maximize fitness gains in simulations when beneficial mutations are common and clonal interference prevents most of them from fixing. To examine these predictions, we propagated 48 \textit{E. coli} populations with 2-, 8-, 100-, and 1000-fold dilutions for 150 days. Adaptation began earlier and fitness gains were greater with 100- and 1000-fold dilutions than with 8-fold dilutions, consistent with the simulations when beneficial mutations are common. However, the selection pressures in the 2-fold treatment were qualitatively different from the other treatments, violating a critical assumption of the model and simulations. Thus, varying the dilution factor during periodic bottlenecks can have multiple effects on the dynamics of adaptation caused by differential losses of diversity, different numbers of generations, and altered selection.},
keywords = {Descendant Experiments, Fitness Trajectories, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}
2023
Lenski R E
Revisiting the design of the long-term evolution experiment with Escherichia coli Journal Article
Journal of Molecular Evolution, vol. 91, no. 3, pp. 241-253, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles
@article{lenski2023,
title = {Revisiting the design of the long-term evolution experiment with \textit{Escherichia coli}},
author = {Richard E. Lenski},
url = {https://link.springer.com/epdf/10.1007/s00239-023-10095-3?sharing_token=zmDHuK0kbvnJBQq1k96fe_e4RwlQNchNByi7wbcMAY53KNkhv6F2YgRIeC8sZGNejxJrvlAGZWInruED5Dqdai5WeU2RAWL2PJNp0pL9QJO39B_ijCtRZcaW8jqM7PclDJfFwL_78U5zNlQYyCOsQwa1Yxha61uXUWhW-Buiq7o=},
doi = {10.1007/s00239-023-10095-3},
year = {2023},
date = {2023-06-01},
urldate = {2023-02-15},
journal = {Journal of Molecular Evolution},
volume = {91},
number = {3},
pages = {241-253},
abstract = {The long-term evolution experiment (LTEE) with \textit{Escherichia coli} began in 1988 and it continues to this day, with its 12 populations having recently reached 75,000 generations of evolution in a simple, well-controlled environment. The LTEE was designed to explore open-ended questions about the dynamics and repeatability of phenotypic and genetic evolution. Here I discuss various aspects of the LTEE’s experimental design that have enabled its stability and success, including the choices of the culture regime, growth medium, ancestral strain, and statistical replication. I also discuss some of the challenges associated with a long-running project, such as handling procedural errors (e.g., cross-contamination) and managing the expanding collection of frozen samples. The simplicity of the experimental design and procedures have supported the long-term stability of the LTEE. That stability—along with the inherent creativity of the evolutionary process and the emergence of new genomic technologies—provides a platform that has allowed talented students and collaborators to pose questions, collect data, and make discoveries that go far beyond anything I could have imagined at the start of the LTEE.},
keywords = {Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles},
pubstate = {published},
tppubtype = {article}
}
Ascensao J A; Wetmore K M; Good B H; Arkin A P; Hallatschek O
Quantifying the adaptive potential of a nascent bacterial community Journal Article
Nature Communications, vol. 14, no. 1, pp. 248, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Descendant Experiments, Genome Evolution, Genotypes and Phenotypes
@article{ascensao2023,
title = {Quantifying the adaptive potential of a nascent bacterial community},
author = {Joao A. Ascensao and Kelly M. Wetmore and Benjamin H. Good and Adam P. Arkin and Oskar Hallatschek},
url = {https://www.nature.com/articles/s41467-022-35677-5},
doi = {10.1038/s41467-022-35677-5},
year = {2023},
date = {2023-01-16},
urldate = {2022-01-01},
journal = {Nature Communications},
volume = {14},
number = {1},
pages = {248},
publisher = {Cold Spring Harbor Laboratory},
abstract = {The fitness effects of all possible mutations available to an organism largely shape the dynamics of evolutionary adaptation. Yet, whether and how this adaptive landscape changes over evolutionary times, especially upon ecological diversification and changes in community composition, remains poorly understood. We sought to fill this gap by analyzing a stable community of two closely related ecotypes (“L” and “S”) shortly after they emerged within the \textit{E. coli} Long-Term Evolution Experiment (LTEE). We engineered genome-wide barcoded transposon libraries to measure the invasion fitness effects of all possible gene knockouts in the coexisting strains as well as their ancestor, for many different, ecologically relevant conditions. We find consistent statistical patterns of fitness effect variation across both genetic background and community composition, despite the idiosyncratic behavior of individual knockouts. Additionally, fitness effects are correlated with evolutionary outcomes for a number of conditions, possibly revealing shifting patterns of adaptation. Together, our results reveal how ecological and epistatic effects combine to shape the adaptive landscape in a nascent ecological community.},
keywords = {Demography and Ecology, Descendant Experiments, Genome Evolution, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2022
Izutsu M; Lenski R E
Experimental test of the contributions of initial variation and new mutations to adaptive evolution in a novel environment Journal Article
Frontiers in Ecology and Evolution, vol. 10, pp. 958406, 2022, ISSN: 2296-701X.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Historical Contingency, Parallelism and Divergence
@article{izutsu2022,
title = {Experimental test of the contributions of initial variation and new mutations to adaptive evolution in a novel environment},
author = {Minako Izutsu and Richard E. Lenski},
url = {https://www.frontiersin.org/articles/10.3389/fevo.2022.958406/full},
doi = {10.3389/fevo.2022.958406},
issn = {2296-701X},
year = {2022},
date = {2022-10-06},
urldate = {2022-10-01},
journal = {Frontiers in Ecology and Evolution},
volume = {10},
pages = {958406},
abstract = {Experimental evolution is an approach that allows researchers to study organisms as they evolve in controlled environments. Despite the growing popularity of this approach, there are conceptual gaps among projects that use different experimental designs. One such gap concerns the contributions to adaptation of genetic variation present at the start of an experiment and that of new mutations that arise during an experiment. The primary source of genetic variation has historically depended largely on the study organisms. In the long-term evolution experiment (LTEE) using \textit{Escherichia coli}, for example, each population started from a single haploid cell, and therefore, adaptation depended entirely on new mutations. Most other microbial evolution experiments have followed the same strategy. By contrast, evolution experiments using multicellular, sexually reproducing organisms typically start with preexisting variation that fuels the response to selection. New mutations may also come into play in later generations of these experiments, but it is generally difficult to quantify their contribution in these studies. Here, we performed an experiment using \textit{E. coli} to compare the contributions of initial genetic variation and new mutations to adaptation in a new environment. Our experiment had four treatments that varied in their starting diversity, with 18 populations in each treatment. One treatment depended entirely on new mutations, while the other three began with mixtures of clones, whole-population samples, or mixtures of whole-population samples from the LTEE. We tracked a genetic marker associated with different founders in two treatments. These data revealed significant variation in fitness among the founders, and that variation impacted evolution in the early generations of our experiment. However, there were no differences in fitness among the treatments after 500 or 2,000 generations in the new environment, despite the variation in fitness among the founders. These results indicate that new mutations quickly dominated, and eventually they contributed more to adaptation than did the initial variation. Our study thus shows that preexisting genetic variation can have a strong impact on early evolution in a new environment, but new beneficial mutations may contribute more to later evolution and can even drive some initially beneficial variants to extinction.},
keywords = {Descendant Experiments, Historical Contingency, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
Smith C E; Smith A N H; Cooper T F; Moore F B -G
Fitness of evolving bacterial populations is contingent on deep and shallow history but only shallow history creates predictable patterns Journal Article
Proceedings of the Royal Society B: Biological Sciences, vol. 289, no. 1982, pp. 20221292, 2022.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Genotypes and Phenotypes, Historical Contingency
@article{smith2022,
title = {Fitness of evolving bacterial populations is contingent on deep and shallow history but only shallow history creates predictable patterns},
author = {Chelsea E. Smith AND Adam N. H. Smith AND Tim F. Cooper AND Francisco B.-G. Moore},
url = {https://royalsocietypublishing.org/doi/10.1098/rspb.2022.1292},
doi = {10.1098/rspb.2022.1292},
year = {2022},
date = {2022-09-14},
journal = {Proceedings of the Royal Society B: Biological Sciences},
volume = {289},
number = {1982},
pages = {20221292},
abstract = {Long-term evolution experiments have tested the importance of genetic and environmental factors in influencing evolutionary outcomes. Differences in phylogenetic history, recent adaptation to distinct environments and chance events, all influence the fitness of a population. However, the interplay of these factors on a population's evolutionary potential remains relatively unexplored. We tracked the outcome of 2000 generations of evolution of four natural isolates of \textit{Escherichia coli} bacteria that were engineered to also create differences in shallow history by adding previously identified mutations selected in a separate long-term experiment. Replicate populations started from each progenitor evolved in four environments. We found that deep and shallow phylogenetic histories both contributed significantly to differences in evolved fitness, though by different amounts in different selection environments. With one exception, chance effects were not significant. Whereas the effect of deep history did not follow any detectable pattern, effects of shallow history followed a pattern of diminishing returns whereby fitter ancestors had smaller fitness increases. These results are consistent with adaptive evolution being contingent on the interaction of several evolutionary forces but demonstrate that the nature of these interactions is not fixed and may not be predictable even when the role of chance is small.},
keywords = {Descendant Experiments, Fitness Trajectories, Genotypes and Phenotypes, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Jordan J A; Lenski R E; Card K J
Idiosyncratic fitness costs of ampicillin-resistant mutants derived from a long-term experiment with Escherichia coli Journal Article
Antibiotics, vol. 11, no. 3, pp. 347, 2022, ISSN: 2079-6382.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution, Historical Contingency
@article{Jordan2022.02.06.479266,
title = {Idiosyncratic fitness costs of ampicillin-resistant mutants derived from a long-term experiment with \textit{Escherichia coli}},
author = {Jalin A. Jordan and Richard E. Lenski and Kyle J. Card},
url = {https://www.biorxiv.org/content/10.1101/2022.02.06.479266v1},
doi = {10.3390/antibiotics11030347},
issn = {2079-6382},
year = {2022},
date = {2022-03-13},
urldate = {2022-03-13},
journal = {Antibiotics},
volume = {11},
number = {3},
pages = {347},
publisher = {Cold Spring Harbor Laboratory},
abstract = {Antibiotic resistance is a growing concern that has prompted a renewed focus on drug discovery, stewardship, and evolutionary studies of the patterns and processes that underlie this phenomenon. A resistant strain’s competitive fitness relative to its sensitive counterparts in the absence of drug can impact its spread and persistence in both clinical and community settings. In a prior study, we examined the fitness of tetracycline-resistant clones that evolved from five different Escherichia coli genotypes, which had diverged during a long-term evolution experiment. In this study, we build on that work to examine whether ampicillin-resistant mutants are also less fit in the absence of the drug than their sensitive parents, and whether the cost of resistance is constant or variable among independently derived lines. Like the tetracycline-resistant lines, the ampicillin-resistant mutants were often less fit than their sensitive parents, with significant variation in the fitness costs among the mutants. This variation was not associated with the level of resistance conferred by the mutations, nor did it vary across the different parental backgrounds. In our earlier study, some of the variation in fitness costs associated with tetracycline resistance was explained by the effects of different mutations affecting the same cellular pathway and even the same gene. In contrast, the variance among the ampicillin-resistant mutants was associated with different sets of target genes. About half of the resistant clones suffered large fitness deficits, and their mutations impacted major outer-membrane proteins or subunits of RNA polymerases. The other mutants experienced little or no fitness costs and with, one exception, they had mutations affecting other genes and functions. Our findings underscore the importance of comparative studies on the evolution of antibiotic resistance, and they highlight the nuanced processes that shape these phenotypes.},
keywords = {Descendant Experiments, Genome Evolution, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
2021
Deatherage D E; Barrick J E
High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing Journal Article
Cell Systems, vol. 12, no. 12, pp. 1187-1200, 2021, ISSN: 24054712.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous
@article{DEATHERAGE2021,
title = {High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing},
author = {Daniel E. Deatherage and Jeffrey E. Barrick},
url = {https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8678185/},
doi = {10.1016/j.cels.2021.08.011},
issn = {24054712},
year = {2021},
date = {2021-09-01},
urldate = {2021-09-01},
journal = {Cell Systems},
volume = {12},
number = {12},
pages = {1187-1200},
abstract = {Understanding how cells are likely to evolve can guide medical interventions and bioengineering efforts that must contend with unwanted mutations. The adaptome of a cell—the neighborhood of genetic changes that are most likely to drive adaptation in a given environment—can be mapped by tracking rare beneficial variants during the early stages of clonal evolution. We used multiplex adaptome capture sequencing (mAdCap-seq), a procedure that combines unique molecular identifiers and hybridization-based enrichment, to characterize mutations in eight \textit{Escherichia coli} genes known to be under selection in a laboratory environment. We tracked 301 mutations at frequencies as low as 0.01% and inferred the fitness effects of 240 of these mutations. There were distinct molecular signatures of selection on protein structure and function for the three genes with the most beneficial mutations. Our results demonstrate how mAdCap-seq can be used to deeply profile a targeted portion of a cell's adaptome.},
keywords = {Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
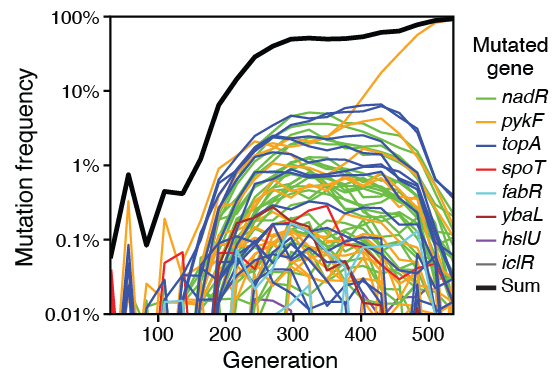
van Raay K; Stolyar S; Sevigny J; Draghi J; Lenski R E; Marx C J; Kerr B; Zaman L
Evolution with private resources reverses some changes from long-term evolution with public resources Unpublished
bioRxiv, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous
@unpublished{Raay2021,
title = {Evolution with private resources reverses some changes from long-term evolution with public resources},
author = {Katrina {van Raay} and Sergey Stolyar and Jordana Sevigny and Jeremy Draghi and Richard E. Lenski and Christopher J. Marx and Benjamin Kerr and Luis Zaman},
url = {https://www.biorxiv.org/content/10.1101/2021.07.11.451942v1},
doi = {https://doi.org/10.1101/2021.07.11.451942},
year = {2021},
date = {2021-07-12},
urldate = {2021-07-12},
journal = {bioRxiv},
pages = {2021.07.11.451942},
abstract = {A population under selection to improve one trait may evolve a sub-optimal state for another trait due to tradeoffs and other evolutionary constraints. How this evolution affects the capacity of a population to adapt when conditions change to favor the second trait is an open question. We investigated this question using isolates from a lineage spanning 60,000 generations of the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}, where cells have access to a shared pool of resources, and have evolved increased competitive ability and a concomitant reduction in numerical yield. Using media-in oil emulsions we shifted the focus of selection to numerical yield, where cells grew in isolated patches with private resources. We found that the time spent evolving under shared resources did not affect the ability to re-evolve toward higher numerical yield. The evolution of numerical yield commonly occurred through mutations in the phosphoenolpyruvate phosphotransferase system. These mutants exhibit slower uptake of glucose, making them poorer competitors for public resources, and produce smaller cells that release less carbon as overflow metabolites. Our results demonstrate that mutations that were not part of adaptation under one selective regime may enable access to ancestral phenotypes when selection changes to favor evolutionary reversion. },
howpublished = {bioRxiv},
keywords = {Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {unpublished}
}
Karkare K; Lai H; Azevedo R B R; Cooper T F
Historical Contingency Causes Divergence in Adaptive Expression of the lac Operon Journal Article
Molecular Biology and Evolution, vol. 38, no. 7, pp. 2869–2879, 2021, ISSN: 1537-1719.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genotypes and Phenotypes
@article{10.1093/molbev/msab077,
title = {Historical Contingency Causes Divergence in Adaptive Expression of the lac Operon},
author = {Kedar Karkare and Huei-Yi Lai and Ricardo B. R. Azevedo and Tim F. Cooper},
editor = {Patricia Wittkopp},
url = {https://academic.oup.com/mbe/article/38/7/2869/6179336},
doi = {10.1093/molbev/msab077},
issn = {1537-1719},
year = {2021},
date = {2021-06-01},
urldate = {2021-06-01},
journal = {Molecular Biology and Evolution},
volume = {38},
number = {7},
pages = {2869--2879},
abstract = {Populations of \textit{Escherichia coli} selected in constant and fluctuating environments containing lactose often adapt by substituting mutations in the lacI repressor that cause constitutive expression of the lac operon. These mutations occur at a high rate and provide a significant benefit. Despite this, eight of 24 populations evolved for 8,000 generations in environments containing lactose contained no detectable repressor mutations. We report here on the basis of this observation. We find that, given relevant mutation rates, repressor mutations are expected to have fixed in all evolved populations if they had maintained the same fitness effect they confer when introduced to the ancestor. In fact, reconstruction experiments demonstrate that repressor mutations have become neutral or deleterious in those populations in which they were not detectable. Populations not fixing repressor mutations nevertheless reached the same fitness as those that did fix them, indicating that they followed an alternative evolutionary path that made redundant the potential benefit of the repressor mutation, but involved unique mutations of equivalent benefit. We identify a mutation occurring in the promoter region of the uspB gene as a candidate for influencing the selective choice between these paths. Our results detail an example of historical contingency leading to divergent evolutionary outcomes.},
keywords = {Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Card K J; Jordan J A; Lenski R E
Idiosyncratic variation in the fitness costs of tetracycline-resistance mutations in Escherichia coli Journal Article
Evolution, vol. 75, no. 5, pp. 1230-1238, 2021, ISSN: 0014-3820.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{nokey,
title = {Idiosyncratic variation in the fitness costs of tetracycline-resistance mutations in \textit{Escherichia coli}},
author = {Kyle J. Card and Jalin A. Jordan and Richard E. Lenski},
url = {https://onlinelibrary.wiley.com/doi/10.1111/evo.14203},
doi = {10.1111/evo.14203},
issn = {0014-3820},
year = {2021},
date = {2021-02-25},
urldate = {2021-02-25},
journal = {Evolution},
volume = {75},
number = {5},
pages = {1230-1238},
abstract = {A bacterium's fitness relative to its competitors, both in the presence and absence of antibiotics, plays a key role in its ecological success and clinical impact. In this study, we examine whether tetracycline-resistant mutants are less fit in the absence of the drug than their sensitive parents, and whether the fitness cost of resistance is constant or variable across independently derived lines. Tetracycline-resistant lines suffered, on average, a reduction in fitness of almost 8%. There was substantial among-line variation in the fitness cost. This variation was not associated with the level of resistance conferred by the mutations, nor did it vary significantly across several genetic backgrounds. The two resistant lines with the most extreme fitness costs involved functionally unrelated mutations on different genetic backgrounds. However, there was also significant variation in the fitness costs for mutations affecting the same pathway and even different alleles of the same gene. Our findings demonstrate that the fitness costs of antibiotic resistance do not always correlate with the phenotypic level of resistance or the underlying genetic changes. Instead, these costs reflect the idiosyncratic effects of particular resistance mutations and the genetic backgrounds in which they occur.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
Card K J; Thomas M D; Graves J L; Barrick J E; Lenski R E
Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with Escherichia coli Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 118, no. 5, pp. e2016886118, 2021, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution, Historical Contingency
@article{Card2021,
title = {Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with \textit{Escherichia coli}},
author = {Kyle J. Card and Misty D. Thomas and Joseph L. Graves and Jeffrey E. Barrick and Richard E. Lenski},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.2016886118},
doi = {10.1073/pnas.2016886118},
issn = {0027-8424},
year = {2021},
date = {2021-02-01},
urldate = {2021-02-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {118},
number = {5},
pages = {e2016886118},
abstract = {Antibiotic resistance is a growing health concern. Efforts to control resistance would benefit from an improved ability to forecast when and how it will evolve. Epistatic interactions between mutations can promote divergent evolutionary trajectories, which complicates our ability to predict evolution. We recently showed that differences between genetic backgrounds can lead to idiosyncratic responses in the evolvability of phenotypic resistance, even among closely related \textit{Escherichia coli} strains. In this study, we examined whether a strain's genetic background also influences the genotypic evolution of resistance. Do lineages founded by different genotypes take parallel or divergent mutational paths to achieve their evolved resistance states? We addressed this question by sequencing the complete genomes of antibiotic-resistant clones that evolved from several different genetic starting points during our earlier experiments. We first validated our statistical approach by quantifying the specificity of genomic evolution with respect to antibiotic treatment. As expected, mutations in particular genes were strongly associated with each drug. Then, we determined that replicate lines evolved from the same founding genotypes had more parallel mutations at the gene level than lines evolved from different founding genotypes, although these effects were more subtle than those showing antibiotic specificity. Taken together with our previous work, we conclude that historical contingency can alter both genotypic and phenotypic pathways to antibiotic resistance.},
keywords = {Descendant Experiments, Genome Evolution, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Gifford I; Dasgupta A; Barrick J E
Rates of gene conversions between Escherichia coli ribosomal operons Journal Article
G3: Genes, Genomes, Genetics, vol. 11, no. 2, pp. jkaa002, 2021, ISSN: 21601836.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Mutation Rates
@article{Gifford2021,
title = {Rates of gene conversions between \emph{Escherichia coli} ribosomal operons},
author = {Isaac Gifford and Aurko Dasgupta and Jeffrey E. Barrick},
url = {https://academic.oup.com/g3journal/article/11/2/jkaa002/5974039},
doi = {10.1093/g3journal/jkaa002},
issn = {21601836},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {G3: Genes, Genomes, Genetics},
volume = {11},
number = {2},
pages = {jkaa002},
abstract = {Due to their universal presence and high sequence conservation, ribosomal RNA (rRNA) sequences are used widely in phylogenetics for inferring evolutionary relationships between microbes and in metagenomics for analyzing the composition of microbial communities. Most microbial genomes encode multiple copies of rRNA genes to supply cells with sufficient capacity for protein synthesis. These copies typically undergo concerted evolution that keeps their sequences identical, or nearly so, due to gene conversion, a type of intragenomic recombination that changes one copy of a homologous sequence to exactly match another. Widely varying rates of rRNA gene conversion have previously been estimated by comparative genomics methods and using genetic reporter assays. To more directly measure rates of rRNA intragenomic recombination, we sequenced the seven \textit{Escherichia coli} rRNA operons in 15 lineages that were evolved for ~13,750 generations with frequent single-cell bottlenecks that reduce the effects of selection. We identified 38 gene conversion events and estimated an overall rate of intragenomic recombination within the 16S and 23S genes between rRNA copies of 3.6 × 10^{−4} per genome per generation or 8.6 × 10^{6} per rRNA operon per homologous donor operon per generation. This rate varied only slightly from random expectations at different sites within the rRNA genes and between rRNA operons located at different positions in the genome. Our accurate estimate of the rate of rRNA gene conversions fills a gap in our quantitative understanding of how ribosomal sequences and other multicopy elements diversify and homogenize during microbial genome evolution.},
keywords = {Descendant Experiments, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2020
Blount Z D; Maddamsetti R; Grant N A; Ahmed S T; Jagdish T; Baxter J A; Sommerfeld B A; Tillman A; Moore J; Slonczewski J L; Barrick J E; Lenski R E
Genomic and phenotypic evolution of Escherichia coli in a novel citrate-only resource environment Journal Article
eLife, vol. 9, pp. 1–64, 2020, ISSN: 2050-084X.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes
@article{Blount2020,
title = {Genomic and phenotypic evolution of \textit{Escherichia coli} in a novel citrate-only resource environment},
author = {Zachary D. Blount and Rohan Maddamsetti and Nkrumah A. Grant and Sumaya T. Ahmed and Tanush Jagdish and Jessica A. Baxter and Brooke A. Sommerfeld and Alice Tillman and Jeremy Moore and Joan L. Slonczewski and Jeffrey E. Barrick and Richard E. Lenski},
url = {https://elifesciences.org/articles/55414},
doi = {10.7554/eLife.55414},
issn = {2050-084X},
year = {2020},
date = {2020-05-01},
urldate = {2020-05-01},
journal = {eLife},
volume = {9},
pages = {1--64},
abstract = {Evolutionary innovations allow populations to colonize new ecological niches. We previously reported that aerobic growth on citrate (Cit^{+}) evolved in an \textit{Escherichia coli} population during adaptation to a minimal glucose medium containing citrate (DM25). Cit^{+} variants can also grow in citrate-only medium (DM0), a novel environment for \textit{E. coli}. To study adaptation to this niche, we founded two sets of Cit^{+} populations and evolved them for 2500 generations in DM0 or DM25. The evolved lineages acquired numerous parallel mutations, many mediated by transposable elements. Several also evolved amplifications of regions containing the \textit{maeA} gene. Unexpectedly, some evolved populations and clones show apparent declines in fitness. We also found evidence of substantial cell death in Cit^{+} clones. Our results thus demonstrate rapid trait refinement and adaptation to the new citrate niche, while also suggesting a recalcitrant mismatch between \textit{E. coli} physiology and growth on citrate.},
keywords = {Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2019
Card K J; LaBar T; Gomez J B; Lenski R E
Historical contingency in the evolution of antibiotic resistance after decades of relaxed selection Journal Article
PLOS Biology, vol. 17, no. 10, pp. e3000397, 2019, ISSN: 1545-7885.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Descendant Experiments, Historical Contingency
@article{nokey,
title = {Historical contingency in the evolution of antibiotic resistance after decades of relaxed selection},
author = {Kyle J. Card and Thomas LaBar and Jasper B. Gomez and Richard E. Lenski},
url = {https://dx.plos.org/10.1371/journal.pbio.3000397},
doi = {10.1371/journal.pbio.3000397},
issn = {1545-7885},
year = {2019},
date = {2019-10-23},
urldate = {2019-10-23},
journal = {PLOS Biology},
volume = {17},
number = {10},
pages = {e3000397},
abstract = {Populations often encounter changed environments that remove selection for the maintenance of particular phenotypic traits. The resulting genetic decay of those traits under relaxed selection reduces an organism’s fitness in its prior environment. However, whether and how such decay alters the subsequent evolvability of a population upon restoration of selection for a previously diminished trait is not well understood. We addressed this question using \textit{Escherichia coli} strains from the long-term evolution experiment (LTEE) that independently evolved for multiple decades in the absence of antibiotics. We first confirmed that these derived strains are typically more sensitive to various antibiotics than their common ancestor. We then subjected the ancestral and derived strains to various concentrations of these drugs to examine their potential to evolve increased resistance. We found that evolvability was idiosyncratic with respect to initial genotype; that is, the derived strains did not generally compensate for their greater susceptibility by “catching up” to the resistance level of the ancestor. Instead, the capacity to evolve increased resistance was constrained in some backgrounds, implying that evolvability depended upon prior mutations in a historically contingent fashion. We further subjected a time series of clones from one LTEE population to tetracycline and determined that an evolutionary constraint arose early in that population, corroborating the role of contingency. In summary, relaxed selection not only can drive populations to increased antibiotic susceptibility, but it can also affect the subsequent evolvability of antibiotic resistance in an unpredictable manner. This conclusion has potential implications for public health, and it underscores the need to consider the genetic context of pathogens when designing drug-treatment strategies.},
keywords = {Correlated Responses, Descendant Experiments, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Lamrabet O; Plumbridge J; Martin M; Lenski R E; Schneider D; Hindré T
Plasticity of Promoter-Core Sequences Allows Bacteria to Compensate for the Loss of a Key Global Regulatory Gene Journal Article
Molecular Biology and Evolution, vol. 36, no. 6, pp. 1121–1133, 2019, ISSN: 0737-4038.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{Lamrabet2019b,
title = {Plasticity of Promoter-Core Sequences Allows Bacteria to Compensate for the Loss of a Key Global Regulatory Gene},
author = {Otmane Lamrabet and Jacqueline Plumbridge and Mikaël Martin and Richard E. Lenski and Dominique Schneider and Thomas Hindré},
editor = {Csaba Pal},
url = {https://academic.oup.com/mbe/article/36/6/1121/5368491},
doi = {10.1093/molbev/msz042},
issn = {0737-4038},
year = {2019},
date = {2019-06-01},
urldate = {2019-06-01},
journal = {Molecular Biology and Evolution},
volume = {36},
number = {6},
pages = {1121--1133},
abstract = {Transcription regulatory networks (TRNs) are of central importance for both short-term phenotypic adaptation in response to environmental fluctuations and long-term evolutionary adaptation, with global regulatory genes often being targets of natural selection in laboratory experiments. Here, we combined evolution experiments, whole-genome resequencing, and molecular genetics to investigate the driving forces, genetic constraints, and molecular mechanisms that dictate how bacteria can cope with a drastic perturbation of their TRNs. The \textit{crp} gene, encoding a major global regulator in \textit{Escherichia coli}, was deleted in four different genetic backgrounds, all derived from the Long-Term Evolution Experiment (LTEE) but with different TRN architectures. We confirmed that \textit{crp} deletion had a more deleterious effect on growth rate in the LTEE-adapted genotypes; and we showed that the \textit{ptsG} gene, which encodes the major glucose-PTS transporter, gained CRP (cyclic AMP receptor protein) dependence over time in the LTEE. We then further evolved the four \textit{crp}-deleted genotypes in glucose minimal medium, and we found that they all quickly recovered from their growth defects by increasing glucose uptake. We showed that this recovery was specific to the selective environment and consistently relied on mutations in the cis-regulatory region of \textit{ptsG}, regardless of the initial genotype. These mutations affected the interplay of transcription factors acting at the promoters, changed the intrinsic properties of the existing promoters, or produced new transcription initiation sites. Therefore, the plasticity of even a single promoter region can compensate by three different mechanisms for the loss of a key regulatory hub in the \textit{E. coli} TRN.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
2018
Maddamsetti R; Lenski R E
PLOS Genetics, vol. 14, no. 1, pp. e1007199, 2018, ISSN: 1553-7404.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution
@article{Maddamsetti2018,
title = {Analysis of bacterial genomes from an evolution experiment with horizontal gene transfer shows that recombination can sometimes overwhelm selection},
author = {Rohan Maddamsetti and Richard E. Lenski},
editor = {Ivan Matic},
url = {https://dx.plos.org/10.1371/journal.pgen.1007199},
doi = {10.1371/journal.pgen.1007199},
issn = {1553-7404},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {PLOS Genetics},
volume = {14},
number = {1},
pages = {e1007199},
abstract = {Few experimental studies have examined the role that sexual recombination plays in bacterial evolution, including the effects of horizontal gene transfer on genome structure. To address this limitation, we analyzed genomes from an experiment in which \textit{Escherichia coli} K-12 Hfr (high frequency recombination) donors were periodically introduced into 12 evolving populations of \textit{E. coli} B and allowed to conjugate repeatedly over the course of 1000 generations. Previous analyses of the evolved strains from this experiment showed that recombination did not accelerate adaptation, despite increasing genetic variation relative to asexual controls. However, the resolution in that previous work was limited to only a few genetic markers. We sought to clarify and understand these puzzling results by sequencing complete genomes from each population. The effects of recombination were highly variable: one lineage was mostly derived from the donors, while another acquired almost no donor DNA. In most lineages, some regions showed repeated introgression and others almost none. Regions with high introgression tended to be near the donors' origin of transfer sites. To determine whether introgressed alleles imposed a genetic load, we extended the experiment for 200 generations without recombination and sequenced whole-population samples. Beneficial alleles in the recipient populations were occasionally driven extinct by maladaptive donor-derived alleles. On balance, our analyses indicate that the plasmid-mediated recombination was sufficiently frequent to drive donor alleles to fixation without providing much, if any, selective advantage.},
keywords = {Descendant Experiments, Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
2017
Deatherage D E; Kepner J L; Bennett A F; Lenski R E; Barrick J E
Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 114, no. 10, pp. E1904–E1912, 2017, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{Deatherage2017,
title = {Specificity of genome evolution in experimental populations of \textit{Escherichia coli} evolved at different temperatures},
author = {Daniel E. Deatherage and Jamie L. Kepner and Albert F. Bennett and Richard E. Lenski and Jeffrey E. Barrick},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.1616132114},
doi = {10.1073/pnas.1616132114},
issn = {0027-8424},
year = {2017},
date = {2017-03-01},
urldate = {2017-03-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {114},
number = {10},
pages = {E1904--E1912},
abstract = {Isolated populations derived from a common ancestor are expected to diverge genetically and phenotypically as they adapt to different local environments. To examine this process, 30 populations of \textit{Escherichia coli} were evolved for 2,000 generations, with six in each of five different thermal regimes: constant 20 °C, 32 °C, 37 °C, 42 °C, and daily alternations between 32 °C and 42 °C. Here, we sequenced the genomes of one endpoint clone from each population to test whether the history of adaptation in different thermal regimes was evident at the genomic level. The evolved strains had accumulated ∼5.3 mutations, on average, and exhibited distinct signatures of adaptation to the different environments. On average, two strains that evolved under the same regime exhibited ∼17% overlap in which genes were mutated, whereas pairs that evolved under different conditions shared only ∼4%. For example, all six strains evolved at 32 °C had mutations in nadR , whereas none of the other 24 strains did. However, a population evolved at 37 °C for an additional 18,000 generations eventually accumulated mutations in the signature genes strongly associated with adaptation to the other temperature regimes. Two mutations that arose in one temperature treatment tended to be beneficial when tested in the others, although less so than in the regime in which they evolved. These findings demonstrate that genomic signatures of adaptation can be highly specific, even with respect to subtle environmental differences, but that this imprint may become obscured over longer timescales as populations continue to change and adapt to the shared features of their environments.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
2015
Satterwhite R S; Cooper T F
Constraints on adaptation of Escherichia coli to mixed-resource environments increase over time Journal Article
Evolution, vol. 69, no. 8, pp. 2067–2078, 2015, ISSN: 00143820.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{Satterwhite2015,
title = {Constraints on adaptation of \textit{Escherichia coli} to mixed-resource environments increase over time},
author = {Rebecca S. Satterwhite and Tim F. Cooper},
url = {https://onlinelibrary.wiley.com/doi/10.1111/evo.12710},
doi = {10.1111/evo.12710},
issn = {00143820},
year = {2015},
date = {2015-08-01},
urldate = {2015-08-01},
journal = {Evolution},
volume = {69},
number = {8},
pages = {2067--2078},
abstract = {Can a population evolved in two resources reach the same fitness in both as specialist populations evolved in each of the individual resources? This question is central to theories of ecological specialization, the maintenance of genetic variation, and sympatric speciation, yet relatively few experiments have examined costs of generalism over long-term adaptation. We tested whether selection in environments containing two resources limits a population's ability to adapt to the individual resources by comparing the fitness of replicate \textit{Escherichia coli} populations evolved for 6000 generations in the presence of glucose or lactose alone (specialists), or in varying presentations of glucose and lactose together (generalists). We found that all populations had significant fitness increases in both resources, though the magnitude and rate of these increases differed. For the first 4000 generations, most generalist populations increased in fitness as quickly in the individual resources as the corresponding specialist populations. From 5000 generations, however, a widespread cost of adaptation affected all generalists, indicating a growing constraint on their abilities to adapt to two resources simultaneously. Our results indicate that costs of generalism are prevalent, but may influence evolutionary trajectories only after a period of cost-free adaptation.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}