2023
Barrick J E; Blount Z D; Lake D M; Dwenger J H; Chavarria-Palma J E; Izutsu M; Wiser M J
Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Journal of Visualized Experiments, vol. 198, pp. e65342, 2023, ISSN: 1940-087X.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Methods and Miscellaneous
@article{Barrick2023,
title = {Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli},
author = {Jeffrey E. Barrick and Zachary D. Blount and Devin M. Lake and Jack H. Dwenger and Jesus E. Chavarria-Palma and Minako Izutsu and Michael J. Wiser},
doi = {10.3791/65342},
issn = {1940-087X},
year = {2023},
date = {2023-08-18},
urldate = {2023-08-18},
journal = {Journal of Visualized Experiments},
volume = {198},
pages = {e65342},
abstract = {The Long-Term Evolution Experiment (LTEE) has followed twelve populations of \textit{Escherichia coli} as they have adapted to a simple laboratory environment for more than 35 years and 77,000 bacterial generations. The setup and procedures used in the LTEE epitomize reliable and reproducible methods for studying microbial evolution. In this protocol, we first describe how the LTEE populations are transferred to fresh medium and cultured each day. Then, we describe how the LTEE populations are regularly checked for possible signs of contamination and archived to provide a permanent frozen "fossil record" for later study. Multiple safeguards included in these procedures are designed to prevent contamination, detect various problems when they occur, and recover from disruptions without appreciably setting back the progress of the experiment. One way that the overall tempo and character of evolutionary changes are monitored in the LTEE is by measuring the competitive fitness of populations and strains from the experiment. We describe how co-culture competition assays are conducted and provide both a spreadsheet and an R package (fitnessR) for calculating relative fitness from the results. Over the course of the LTEE, the behaviors of some populations have changed in interesting ways, and new technologies like whole-genome sequencing have provided additional avenues for investigating how the populations have evolved. We end by discussing how the original LTEE procedures have been updated to accommodate or take advantage of these changes. This protocol will be useful for researchers who use the LTEE as a model system for studying connections between evolution and genetics, molecular biology, systems biology, and ecology. More broadly, the LTEE provides a tried-and-true template for those who are beginning their own evolution experiments with new microbes, environments, and questions. },
keywords = {Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
Lenski R E
Revisiting the design of the long-term evolution experiment with Escherichia coli Journal Article
Journal of Molecular Evolution, vol. 91, no. 3, pp. 241-253, 2023.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles
@article{lenski2023,
title = {Revisiting the design of the long-term evolution experiment with \textit{Escherichia coli}},
author = {Richard E. Lenski},
url = {https://link.springer.com/epdf/10.1007/s00239-023-10095-3?sharing_token=zmDHuK0kbvnJBQq1k96fe_e4RwlQNchNByi7wbcMAY53KNkhv6F2YgRIeC8sZGNejxJrvlAGZWInruED5Dqdai5WeU2RAWL2PJNp0pL9QJO39B_ijCtRZcaW8jqM7PclDJfFwL_78U5zNlQYyCOsQwa1Yxha61uXUWhW-Buiq7o=},
doi = {10.1007/s00239-023-10095-3},
year = {2023},
date = {2023-06-01},
urldate = {2023-02-15},
journal = {Journal of Molecular Evolution},
volume = {91},
number = {3},
pages = {241-253},
abstract = {The long-term evolution experiment (LTEE) with \textit{Escherichia coli} began in 1988 and it continues to this day, with its 12 populations having recently reached 75,000 generations of evolution in a simple, well-controlled environment. The LTEE was designed to explore open-ended questions about the dynamics and repeatability of phenotypic and genetic evolution. Here I discuss various aspects of the LTEE’s experimental design that have enabled its stability and success, including the choices of the culture regime, growth medium, ancestral strain, and statistical replication. I also discuss some of the challenges associated with a long-running project, such as handling procedural errors (e.g., cross-contamination) and managing the expanding collection of frozen samples. The simplicity of the experimental design and procedures have supported the long-term stability of the LTEE. That stability—along with the inherent creativity of the evolutionary process and the emergence of new genomic technologies—provides a platform that has allowed talented students and collaborators to pose questions, collect data, and make discoveries that go far beyond anything I could have imagined at the start of the LTEE.},
keywords = {Citrate Evolution, Descendant Experiments, Fitness Trajectories, Genome Evolution, Methods and Miscellaneous, Review Articles},
pubstate = {published},
tppubtype = {article}
}
Jagdish T
The dynamics of fitness and pleiotropy in a long-term evolution experiment with Escherichia coli PhD Thesis
2023.
Abstract | Links | BibTeX | Tags: Correlated Responses, Demography and Ecology, Fitness Trajectories, Methods and Miscellaneous
@phdthesis{nokey,
title = {The dynamics of fitness and pleiotropy in a long-term evolution experiment with \textit{Escherichia coli}},
author = {Tanush Jagdish},
url = {https://dash.harvard.edu/handle/1/37375541},
year = {2023},
date = {2023-05-08},
urldate = {2023-05-08},
abstract = {Life on earth is shaped by a delicate balance between chance and necessity. A combination of natural selection and genetic drift has moulded random genetic variations over the course of billions of years to generate the complex and interconnected biosphere we see today. These evolutionary dynamics are ultimately the product of numerous genetic mechanisms operating in genomes with highly complex architectures. Unravelling the rules of genetic mechanisms responsible for evolutionary innovation is critical to developing a comprehensive and predictive evolutionary theory, but has evaded direct experimentation since the scale of resources and technology needed to study the patterns of genetic evolution has only recently become achievable. In this dissertation, I explore the use of microbial experimental evolution as a powerful tool for probing evolutionary questions, particularly by leveraging DNA-barcoding and high-throughput DNA sequencing. In chapter 1, I provide an overview of the field of microbial experimental evolution, dividing its history into two eras marked by qualitatively different methodological advancements. I make the case that we now stand at the dawn of a third era, where advances in genome-engineering coupled with low-cost, high-throughput DNA sequencing will allow experiments to finally probe evolution on a statistical scale. I then present two research studies that take advantage of microbial experimental evolution to investigate distinct genetic mechanisms key to the evolutionary process. In Chapter 2, I explore whether fitness can continue increasing in a population that has already adapted to its environment for over 30,000 generations, and whether fixations of beneficial mutations from population-wide DNA sequencing can predict jumps in fitness. My findings reveal that fitness continues to monotonically increase in step with the fixation of beneficial mutations, even though the rate of fixation has dramatically slowed down, highlighting the potential for ongoing adaptation even under constant environmental conditions. In Chapter 3, I develop a novel conjugation-based DNA-barcoding method for the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}, allowing me to examine the pleiotropic consequences of adaptation to glucose over 50,000 generations in 15 novel resource environments. My observations reveal broad patterns of both convergent and divergent evolution that correspond with mutations in key metabolic genes in clonal sequencing datasets, shedding light on the nature of pleiotropy and its evolution over extended timescales. Using microbial model systems in an evolutionary context has the unique advantage of being relevant both to fundamental evolutionary biology and human health. Since genetic drift and rare mutational events both play an outsized role in determining the evolutionary trajectories of populations, evolutionary questions in the modern age will increasingly be faced with issues of scale. Microbial experimental evolution offers both scale and tractability to solve this problem. Uniquely, this does not sacrifice on human relevance. Building a coarse-grained and comprehensive evolutionary theory is more significant to society today than ever before as the importance of clonal evolution in cancer, gut microbiomes and even pandemics becomes more clearly understood.},
keywords = {Correlated Responses, Demography and Ecology, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {phdthesis}
}
2021
Deatherage D E; Barrick J E
High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing Journal Article
Cell Systems, vol. 12, no. 12, pp. 1187-1200, 2021, ISSN: 24054712.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous
@article{DEATHERAGE2021,
title = {High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing},
author = {Daniel E. Deatherage and Jeffrey E. Barrick},
url = {https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8678185/},
doi = {10.1016/j.cels.2021.08.011},
issn = {24054712},
year = {2021},
date = {2021-09-01},
urldate = {2021-09-01},
journal = {Cell Systems},
volume = {12},
number = {12},
pages = {1187-1200},
abstract = {Understanding how cells are likely to evolve can guide medical interventions and bioengineering efforts that must contend with unwanted mutations. The adaptome of a cell—the neighborhood of genetic changes that are most likely to drive adaptation in a given environment—can be mapped by tracking rare beneficial variants during the early stages of clonal evolution. We used multiplex adaptome capture sequencing (mAdCap-seq), a procedure that combines unique molecular identifiers and hybridization-based enrichment, to characterize mutations in eight \textit{Escherichia coli} genes known to be under selection in a laboratory environment. We tracked 301 mutations at frequencies as low as 0.01% and inferred the fitness effects of 240 of these mutations. There were distinct molecular signatures of selection on protein structure and function for the three genes with the most beneficial mutations. Our results demonstrate how mAdCap-seq can be used to deeply profile a targeted portion of a cell's adaptome.},
keywords = {Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
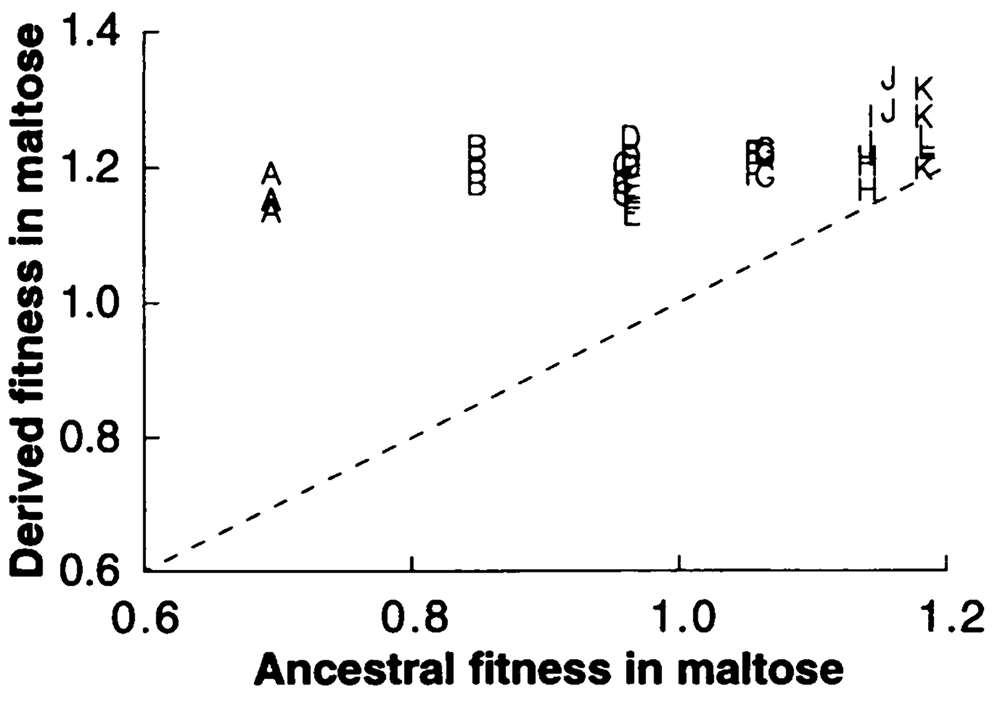
van Raay K; Stolyar S; Sevigny J; Draghi J; Lenski R E; Marx C J; Kerr B; Zaman L
Evolution with private resources reverses some changes from long-term evolution with public resources Unpublished
bioRxiv, 2021.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous
@unpublished{Raay2021,
title = {Evolution with private resources reverses some changes from long-term evolution with public resources},
author = {Katrina {van Raay} and Sergey Stolyar and Jordana Sevigny and Jeremy Draghi and Richard E. Lenski and Christopher J. Marx and Benjamin Kerr and Luis Zaman},
url = {https://www.biorxiv.org/content/10.1101/2021.07.11.451942v1},
doi = {https://doi.org/10.1101/2021.07.11.451942},
year = {2021},
date = {2021-07-12},
urldate = {2021-07-12},
journal = {bioRxiv},
pages = {2021.07.11.451942},
abstract = {A population under selection to improve one trait may evolve a sub-optimal state for another trait due to tradeoffs and other evolutionary constraints. How this evolution affects the capacity of a population to adapt when conditions change to favor the second trait is an open question. We investigated this question using isolates from a lineage spanning 60,000 generations of the Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli}, where cells have access to a shared pool of resources, and have evolved increased competitive ability and a concomitant reduction in numerical yield. Using media-in oil emulsions we shifted the focus of selection to numerical yield, where cells grew in isolated patches with private resources. We found that the time spent evolving under shared resources did not affect the ability to re-evolve toward higher numerical yield. The evolution of numerical yield commonly occurred through mutations in the phosphoenolpyruvate phosphotransferase system. These mutants exhibit slower uptake of glucose, making them poorer competitors for public resources, and produce smaller cells that release less carbon as overflow metabolites. Our results demonstrate that mutations that were not part of adaptation under one selective regime may enable access to ancestral phenotypes when selection changes to favor evolutionary reversion. },
howpublished = {bioRxiv},
keywords = {Cell Morphology, Correlated Responses, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes, Historical Contingency, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {unpublished}
}
2018
Lenski R E; Burnham T C
Experimental evolution of bacteria across 60,000 generations, and what it might mean for economics and human decision-making Journal Article
Journal of Bioeconomics, vol. 20, no. 1, pp. 107–124, 2018, ISSN: 1387-6996.
Abstract | Links | BibTeX | Altmetric | Tags: Methods and Miscellaneous
@article{Lenski2018,
title = {Experimental evolution of bacteria across 60,000 generations, and what it might mean for economics and human decision-making},
author = {Richard E. Lenski and Terence C. Burnham},
url = {http://link.springer.com/10.1007/s10818-017-9258-7},
doi = {10.1007/s10818-017-9258-7},
issn = {1387-6996},
year = {2018},
date = {2018-04-01},
urldate = {2018-04-01},
journal = {Journal of Bioeconomics},
volume = {20},
number = {1},
pages = {107--124},
publisher = {Springer US},
abstract = {Evolutionary biology and economics are both rich in theory and steeped in data, but they also share challenges including the fact that the systems they seek to understand are, in certain respects, unique and not easily manipulated. Nonetheless, both fields have seen growing efforts to provide experimental approaches to address specific issues. Here, we review some results from a 30-year experiment in which 12 populations of bacteria have been evolving for over 60,000 generations to characterize: (i) the time scale of adaptation to new conditions, (ii) the repeatability of evolutionary changes, and (iii) the benefits and costs of specialization. In each case, we speculate on potential connections and implications of these findings for the field of economics. Moreover, both the bacteria in this experiment and people in modern societies live in novel environments, which leads to an evolutionary mismatch between their genes and environments. Regardless of the value of our speculations, we hope this paper stimulates further interest in pursuing experiments in fields that are often viewed as observational and not amenable to experimentation.},
keywords = {Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2015
Wiser M J; Lenski R E
A Comparison of Methods to Measure Fitness in Escherichia coli Journal Article
PLOS ONE, vol. 10, no. 5, pp. e0126210, 2015, ISSN: 1932-6203.
Abstract | Links | BibTeX | Altmetric | Tags: Methods and Miscellaneous
@article{Wiser2015,
title = {A Comparison of Methods to Measure Fitness in \textit{Escherichia coli}},
author = {Michael J. Wiser and Richard E. Lenski},
editor = {Jeffrey L Blanchard},
url = {https://dx.plos.org/10.1371/journal.pone.0126210},
doi = {10.1371/journal.pone.0126210},
issn = {1932-6203},
year = {2015},
date = {2015-05-01},
urldate = {2015-05-01},
journal = {PLOS ONE},
volume = {10},
number = {5},
pages = {e0126210},
abstract = {In order to characterize the dynamics of adaptation, it is important to be able to quantify how a population's mean fitness changes over time. Such measurements are especially important in experimental studies of evolution using microbes. The Long-Term Evolution Experiment (LTEE) with \textit{Escherichia coli} provides one such system in which mean fitness has been measured by competing derived and ancestral populations. The traditional method used to measure fitness in the LTEE and many similar experiments, though, is subject to a potential limitation. As the relative fitness of the two competitors diverges, the measurement error increases because the less-fit population becomes increasingly small and cannot be enumerated as precisely. Here, we present and employ two alternatives to the traditional method. One is based on reducing the fitness differential between the competitors by using a common reference competitor from an intermediate generation that has intermediate fitness; the other alternative increases the initial population size of the less-fit, ancestral competitor. We performed a total of 480 competitions to compare the statistical properties of estimates obtained using these alternative methods with those obtained using the traditional method for samples taken over 50,000 generations from one of the LTEE populations. On balance, neither alternative method yielded measurements that were more precise than the traditional method.},
keywords = {Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2014
Quandt E M; Deatherage D E; Ellington A D; Georgiou G; Barrick J E
Recursive genomewide recombination and sequencing reveals a key refinement step in the evolution of a metabolic innovation in Escherichia coli Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 111, no. 6, pp. 2217–2222, 2014, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Genotypes and Phenotypes, Methods and Miscellaneous
@article{Quandt2014,
title = {Recursive genomewide recombination and sequencing reveals a key refinement step in the evolution of a metabolic innovation in \textit{Escherichia coli}},
author = {Erik M. Quandt and Daniel E. Deatherage and Andrew D. Ellington and George Georgiou and Jeffrey E. Barrick},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.1314561111},
doi = {10.1073/pnas.1314561111},
issn = {0027-8424},
year = {2014},
date = {2014-02-01},
urldate = {2014-02-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {111},
number = {6},
pages = {2217--2222},
abstract = {Evolutionary innovations often arise from complex genetic and ecological interactions, which can make it challenging to understand retrospectively how a novel trait arose. In a long-term experiment, \textit{Escherichia coli} gained the ability to use abundant citrate (Cit+) in the growth medium after ~31,500 generations of evolution. Exploiting this previously untapped resource was highly beneficial: later Cit+ variants achieve a much higher population density in this environment. All Cit+ individuals share a mutation that activates aerobic expression of the \textit{citT} citrate transporter, but this mutation confers only an extremely weak Cit+ phenotype on its own. To determine which of the other >70 mutations in early Cit+ clones were needed to take full advantage of citrate, we developed a recursive genomewide recombination and sequencing method (REGRES) and performed genetic backcrosses to purge mutations not required for Cit+ from an evolved strain. We discovered a mutation that increased expression of the \textit{dctA} C4-dicarboxylate transporter greatly enhanced the Cit+ phenotype after it evolved. Surprisingly, strains containing just the \textit{citT} and \textit{dctA} mutations fully use citrate, indicating that earlier mutations thought to have potentiated the initial evolution of Cit+ are not required for expression of the refined version of this trait. Instead, this metabolic innovation may be contingent on a genetic background, and possibly ecological context, that enabled \textit{citT} mutants to persist among competitors long enough to obtain \textit{dctA} or equivalent mutations that conferred an overwhelming advantage. More generally, refinement of an emergent trait from a rudimentary form may be crucial to its evolutionary success.},
keywords = {Citrate Evolution, Genotypes and Phenotypes, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2009
Daegelen P; Studier F W; Lenski R E; Cure S; Kim J F
Tracing Ancestors and Relatives of Escherichia coli B, and the Derivation of B Strains REL606 and BL21(DE3) Journal Article
Journal of Molecular Biology, vol. 394, no. 4, pp. 634-643, 2009, ISSN: 0022-2836.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Methods and Miscellaneous
@article{DAEGELEN2009634,
title = {Tracing Ancestors and Relatives of \textit{Escherichia coli} B, and the Derivation of B Strains REL606 and BL21(DE3)},
author = {Patrick Daegelen and F. William Studier and Richard E. Lenski and Susan Cure and Jihyun F. Kim},
url = {https://www.sciencedirect.com/science/article/pii/S0022283609011395},
doi = {https://doi.org/10.1016/j.jmb.2009.09.022},
issn = {0022-2836},
year = {2009},
date = {2009-01-01},
urldate = {2009-01-01},
journal = {Journal of Molecular Biology},
volume = {394},
number = {4},
pages = {634-643},
abstract = {Antecedents of \textit{Escherichia coli} B have been traced through publications, inferences, and personal communication to a strain from the Institut Pasteur in Paris used by d'Herelle in his studies of bacteriophages as early as 1918 (a strain not in the current collection). This strain appears to have passed from d'Herelle to Bordet in 1920, and from Bordet to at least three other laboratories by 1925. The strain that Gratia received from Bordet was apparently passed to Bronfenbrenner by 1924 and from him to Luria around 1941. Delbrück and Luria published the first paper calling this strain B in 1942. Its choice as the common host for phages T1–T7 by the phage group that developed around Delbrück, Luria, and Hershey in the 1940s led to widespread use of B along with \textit{E. coli} K-12, chosen about the same time for biochemical and genetic studies by Tatum and Lederberg. Not all currently available strains related to B are descended from the B of Delbrück and Luria; at least three strains with somewhat different characteristics were derived independently by Hershey directly from the Bronfenbrenner strain, and a strain that appears to have passed from Bordet to Wollman is in the current Collection of the Institut Pasteur. The succession of manipulations and strains that led from the B of Delbrück and Luria to REL606 and BL21(DE3) is given, established in part through evidence from their recently determined complete genome sequences.},
keywords = {Genome Evolution, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2002
Schneider D; Duperchy E; Depeyrot J; Coursange E; Lenski R E; Blot M
Genomic comparisons among Escherichia coli strains B, K-12, and O157:H7 using IS elements as molecular markers. Journal Article
BMC microbiology, vol. 2, pp. 18, 2002, ISSN: 1471-2180.
Abstract | Links | BibTeX | Altmetric | Tags: Methods and Miscellaneous
@article{Schneider2002,
title = {Genomic comparisons among \textit{Escherichia coli} strains B, K-12, and O157:H7 using IS elements as molecular markers.},
author = {Dominique Schneider and Esther Duperchy and Joelle Depeyrot and Evelyne Coursange and Richard E. Lenski and Michel Blot},
url = {http://www.ncbi.nlm.nih.gov/pubmed/12106505
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC117601},
doi = {10.1186/1471-2180-2-18},
issn = {1471-2180},
year = {2002},
date = {2002-07-01},
urldate = {2002-07-01},
journal = {BMC microbiology},
volume = {2},
pages = {18},
abstract = {Background
Insertion Sequence (IS) elements are mobile genetic elements widely distributed among bacteria. Their activities cause mutations, promoting genetic diversity and sometimes adaptation. Previous studies have examined their copy number and distribution in \textit{Escherichia coli} K-12 and natural isolates. Here, we map most of the IS elements in \textit{E. coli} B and compare their locations with the published genomes of K-12 and O157:H7.
Results
The genomic locations of IS elements reveal numerous differences between B, K-12, and O157:H7. IS elements occur in \textit{hok-sok} loci (homologous to plasmid stabilization systems) in both B and K-12, whereas these same loci lack IS elements in O157:H7. IS elements in B and K-12 are often found in locations corresponding to O157:H7-specific sequences, which suggests IS involvement in chromosomal rearrangements including the incorporation of foreign DNA. Some sequences specific to B are identified, as reported previously for O157:H7. The extent of nucleotide sequence divergence between B and K-12 is < 2% for most sequences adjacent to IS elements. By contrast, B and K-12 share only a few IS locations besides those in \textit{hok-sok} loci. Several phenotypic features of B are explained by IS elements, including differential porin expression from K-12.
Conclusions
These data reveal a high level of IS activity since \textit{E. coli} B, K-12, and O157:H7 diverged from a common ancestor, including IS association with deletions and incorporation of horizontally acquired genes as well as transpositions. These findings indicate the important role of IS elements in genome plasticity and divergence.},
keywords = {Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
Insertion Sequence (IS) elements are mobile genetic elements widely distributed among bacteria. Their activities cause mutations, promoting genetic diversity and sometimes adaptation. Previous studies have examined their copy number and distribution in Escherichia coli K-12 and natural isolates. Here, we map most of the IS elements in E. coli B and compare their locations with the published genomes of K-12 and O157:H7.
Results
The genomic locations of IS elements reveal numerous differences between B, K-12, and O157:H7. IS elements occur in hok-sok loci (homologous to plasmid stabilization systems) in both B and K-12, whereas these same loci lack IS elements in O157:H7. IS elements in B and K-12 are often found in locations corresponding to O157:H7-specific sequences, which suggests IS involvement in chromosomal rearrangements including the incorporation of foreign DNA. Some sequences specific to B are identified, as reported previously for O157:H7. The extent of nucleotide sequence divergence between B and K-12 is < 2% for most sequences adjacent to IS elements. By contrast, B and K-12 share only a few IS locations besides those in hok-sok loci. Several phenotypic features of B are explained by IS elements, including differential porin expression from K-12.
Conclusions
These data reveal a high level of IS activity since E. coli B, K-12, and O157:H7 diverged from a common ancestor, including IS association with deletions and incorporation of horizontally acquired genes as well as transpositions. These findings indicate the important role of IS elements in genome plasticity and divergence.
1995
Travisano M; Mongold J A; Bennett A F; Lenski R E
Experimental Tests of the Roles of Adaptation, Chance, and History Journal Article
Science, vol. 267, no. January, 1995.
Abstract | Links | BibTeX | Altmetric | Tags: Cell Morphology, Correlated Responses, Descendant Experiments, Fitness Trajectories, Historical Contingency, Methods and Miscellaneous, Parallelism and Divergence
@article{Travisano1995,
title = {Experimental Tests of the Roles of Adaptation, Chance, and History},
author = {Michael Travisano and Judith A. Mongold and Albert F. Bennett and Richard E. Lenski},
url = {https://www.science.org/lookup/doi/10.1126/science.7809610},
doi = {https://doi.org/10.1126/science.7809610},
year = {1995},
date = {1995-01-01},
urldate = {1995-01-01},
journal = {Science},
volume = {267},
number = {January},
abstract = {The contributions of adaptation, chance, and history to the evolution of fitness and cell size were measured in two separate experiments using bacteria. In both experiments, populations propagated in identical environments achieved similar fitnesses, regardless of prior history or subsequent chance events. In contrast, the evolution of cell size, a trait weakly correlated with fitness, was more strongly influenced by history and chance.},
keywords = {Cell Morphology, Correlated Responses, Descendant Experiments, Fitness Trajectories, Historical Contingency, Methods and Miscellaneous, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}