2022
Maddamsetti R; Grant N A
Discovery of positive and purifying selection in metagenomic time series of hypermutator microbial populations Journal Article
PLOS Genetics, vol. 18, no. 8, pp. e1010324, 2022, ISSN: 1553-7404.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{maddamsetti2022,
title = {Discovery of positive and purifying selection in metagenomic time series of hypermutator microbial populations},
author = { Rohan Maddamsetti and Nkrumah A. Grant},
editor = {Jianzhi Zhang},
url = {https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1010324},
doi = {10.1371/journal.pgen.1010324},
issn = {1553-7404},
year = {2022},
date = {2022-08-18},
urldate = {2022-08-18},
journal = {PLOS Genetics},
volume = {18},
number = {8},
pages = {e1010324},
abstract = {A general method to infer both positive and purifying selection during the real-time evolution of hypermutator pathogens would be broadly useful. To this end, we introduce a simple test to infer mode of selection (STIMS) from metagenomic time series of evolving microbial populations. We test STIMS on metagenomic data generated by simulations of bacterial evolution, and on metagenomic data spanning 62,750 generations of Lenski’s long-term evolution experiment with \textit{Escherichia coli} (LTEE). This benchmarking shows that STIMS detects positive selection in both nonmutator and hypermutator populations, and purifying selection in hypermutator populations. Using STIMS, we find strong evidence of ongoing positive selection on key regulators of the \textit{E. coli} gene regulatory network, even in some hypermutator populations. STIMS also detects positive selection on regulatory genes in hypermutator populations of \textit{Pseudomonas aeruginosa} that adapted to subinhibitory concentrations of colistin—an antibiotic of last resort—for just twenty-six days of laboratory evolution. Our results show that the fine-tuning of gene regulatory networks is a general mechanism for rapid and ongoing adaptation. The simplicity of STIMS, together with its intuitive visual interpretation, make it a useful test for positive and purifying selection in metagenomic data sets that track microbial evolution in real-time.},
key = {Maddamsetti2022},
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2021
Consuegra J; Gaffé J; Lenski R E; Hindré T; Barrick J E; Tenaillon O; Schneider D
Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria Journal Article
Nature Communications, vol. 12, no. 1, pp. 980, 2021, ISSN: 2041-1723.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Mutation Rates
@article{Consuegra2021,
title = {Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria},
author = {Jessika Consuegra and Joël Gaffé and Richard E. Lenski and Thomas Hindré and Jeffrey E. Barrick and Olivier Tenaillon and Dominique Schneider},
url = {http://www.nature.com/articles/s41467-021-21210-7},
doi = {https://doi.org/10.1038/s41467-021-21210-7},
issn = {2041-1723},
year = {2021},
date = {2021-12-01},
urldate = {2021-12-01},
journal = {Nature Communications},
volume = {12},
number = {1},
pages = {980},
publisher = {Springer US},
abstract = {Insertion sequences (IS) are ubiquitous bacterial mobile genetic elements, and the mutations they cause can be deleterious, neutral, or beneficial. The long-term dynamics of IS elements and their effects on bacteria are poorly understood, including whether they are primarily genomic parasites or important drivers of adaptation by natural selection. Here, we investigate the dynamics of IS elements and their contribution to genomic evolution and fitness during a long-term experiment with \textit{Escherichia coli}. IS elements account for ~35% of the mutations that reached high frequency through 50,000 generations in those populations that retained the ancestral point-mutation rate. In mutator populations, IS-mediated mutations are only half as frequent in absolute numbers. In one population, an exceptionally high ~8-fold increase in IS 150 copy number is associated with the beneficial effects of early insertion mutations; however, this expansion later slowed down owing to reduced IS 150 activity. This population also achieves the lowest fitness, suggesting that some avenues for further adaptation are precluded by the IS 150 -mediated mutations. More generally, across all populations, we find that higher IS activity becomes detrimental to adaptation over evolutionary time. Therefore, IS-mediated mutations can both promote and constrain evolvability.},
keywords = {Fitness Trajectories, Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
Gifford I; Dasgupta A; Barrick J E
Rates of gene conversions between Escherichia coli ribosomal operons Journal Article
G3: Genes, Genomes, Genetics, vol. 11, no. 2, pp. jkaa002, 2021, ISSN: 21601836.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Mutation Rates
@article{Gifford2021,
title = {Rates of gene conversions between \emph{Escherichia coli} ribosomal operons},
author = {Isaac Gifford and Aurko Dasgupta and Jeffrey E. Barrick},
url = {https://academic.oup.com/g3journal/article/11/2/jkaa002/5974039},
doi = {10.1093/g3journal/jkaa002},
issn = {21601836},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {G3: Genes, Genomes, Genetics},
volume = {11},
number = {2},
pages = {jkaa002},
abstract = {Due to their universal presence and high sequence conservation, ribosomal RNA (rRNA) sequences are used widely in phylogenetics for inferring evolutionary relationships between microbes and in metagenomics for analyzing the composition of microbial communities. Most microbial genomes encode multiple copies of rRNA genes to supply cells with sufficient capacity for protein synthesis. These copies typically undergo concerted evolution that keeps their sequences identical, or nearly so, due to gene conversion, a type of intragenomic recombination that changes one copy of a homologous sequence to exactly match another. Widely varying rates of rRNA gene conversion have previously been estimated by comparative genomics methods and using genetic reporter assays. To more directly measure rates of rRNA intragenomic recombination, we sequenced the seven \textit{Escherichia coli} rRNA operons in 15 lineages that were evolved for ~13,750 generations with frequent single-cell bottlenecks that reduce the effects of selection. We identified 38 gene conversion events and estimated an overall rate of intragenomic recombination within the 16S and 23S genes between rRNA copies of 3.6 × 10^{−4} per genome per generation or 8.6 × 10^{6} per rRNA operon per homologous donor operon per generation. This rate varied only slightly from random expectations at different sites within the rRNA genes and between rRNA operons located at different positions in the genome. Our accurate estimate of the rate of rRNA gene conversions fills a gap in our quantitative understanding of how ribosomal sequences and other multicopy elements diversify and homogenize during microbial genome evolution.},
keywords = {Descendant Experiments, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2020
Maddamsetti R; Grant N A
Divergent Evolution of Mutation Rates and Biases in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Genome Biology and Evolution, vol. 12, no. 9, pp. 1591-1603, 2020, ISSN: 1759-6653.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{nokey,
title = {Divergent Evolution of Mutation Rates and Biases in the Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Rohan Maddamsetti and Nkrumah A. Grant},
editor = {George Zhang},
url = {https://academic.oup.com/gbe/article/12/9/1591/5898197},
doi = {10.1093/gbe/evaa178},
issn = {1759-6653},
year = {2020},
date = {2020-08-21},
urldate = {2020-08-21},
journal = {Genome Biology and Evolution},
volume = {12},
number = {9},
pages = {1591-1603},
abstract = {All organisms encode enzymes that replicate, maintain, pack, recombine, and repair their genetic material. For this reason, mutation rates and biases also evolve by mutation, variation, and natural selection. By examining metagenomic time series of the Lenski long-term evolution experiment (LTEE) with \textit{Escherichia coli} (Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. 2017. The dynamics of molecular evolution over 60,000 generations. Nature 551(7678):45–50.), we find that local mutation rate variation has evolved during the LTEE. Each LTEE population has evolved idiosyncratic differences in their rates of point mutations, indels, and mobile element insertions, due to the fixation of various hypermutator and antimutator alleles. One LTEE population, called Ara+3, shows a strong, symmetric wave pattern in its density of point mutations, radiating from the origin of replication. This pattern is largely missing from the other LTEE populations, most of which evolved missense, indel, or structural mutations in \textit{topA}, \textit{fis}, and \textit{dusB}—loci that all affect DNA topology. The distribution of mutations in those genes over time suggests epistasis and historical contingency in the evolution of DNA topology, which may have in turn affected local mutation rates. Overall, the replicate populations of the LTEE have largely diverged in their mutation rates and biases, even though they have adapted to identical abiotic conditions.},
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2017
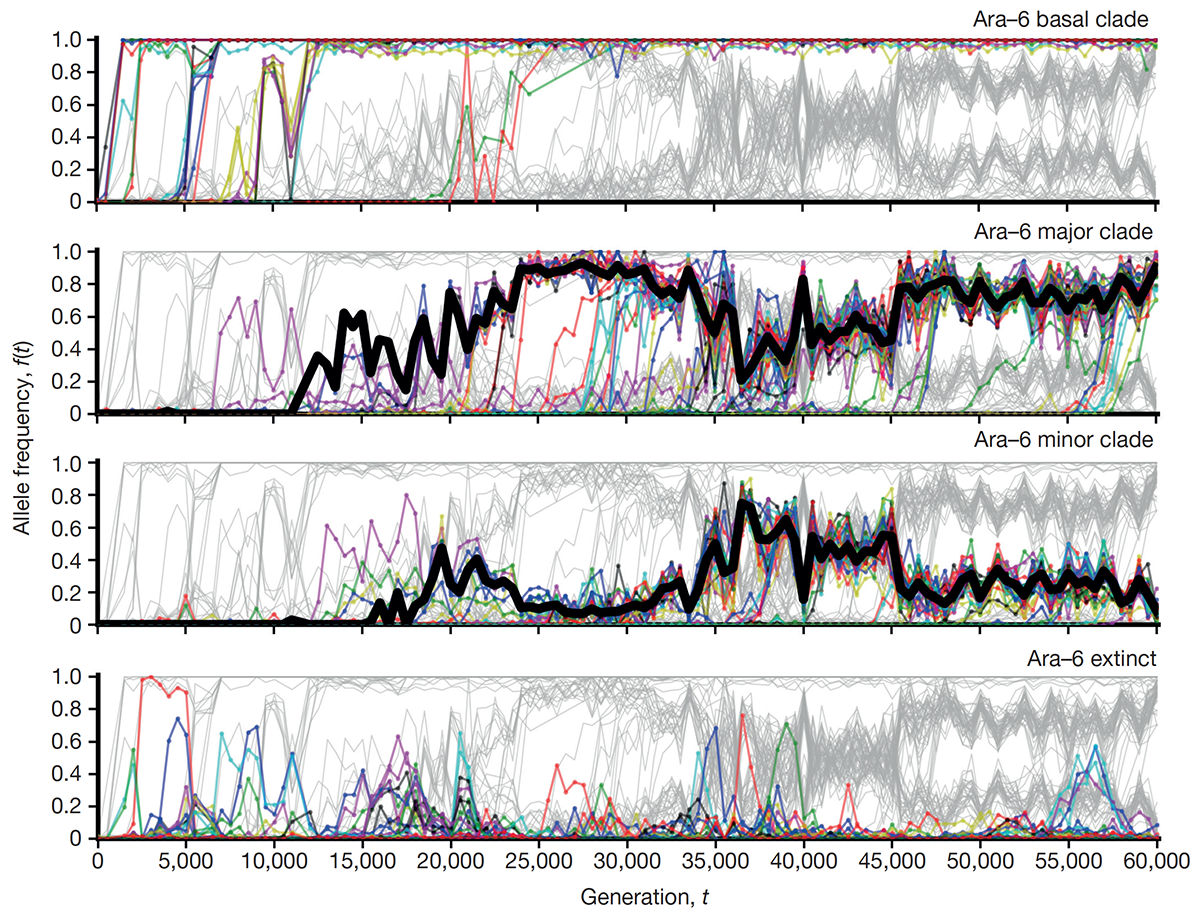
Good B H; McDonald M J; Barrick J E; Lenski R E; Desai M M
The dynamics of molecular evolution over 60,000 generations Journal Article
Nature, vol. 551, no. 7678, pp. 45–50, 2017, ISSN: 14764687.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Genome Evolution, Historical Contingency, Mutation Rates, Parallelism and Divergence
@article{Good2017,
title = {The dynamics of molecular evolution over 60,000 generations},
author = {Benjamin H. Good and Michael J. McDonald and Jeffrey E. Barrick and Richard E. Lenski and Michael M. Desai},
url = {http://dx.doi.org/10.1038/nature24287},
doi = {10.1038/nature24287},
issn = {14764687},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Nature},
volume = {551},
number = {7678},
pages = {45--50},
publisher = {Nature Publishing Group},
abstract = {The outcomes of evolution are determined by a stochastic dynamical process that governs how mutations arise and spread through a population. However, it is difficult to observe these dynamics directly over long periods and across entire genomes. Here we analyse the dynamics of molecular evolution in twelve experimental populations of \textit{Escherichia coli}, using whole-genome metagenomic sequencing at five hundred-generation intervals through sixty thousand generations. Although the rate of fitness gain declines over time, molecular evolution is characterized by signatures of rapid adaptation throughout the duration of the experiment, with multiple beneficial variants simultaneously competing for dominance in each population. Interactions between ecological and evolutionary processes play an important role, as long-term quasi-stable coexistence arises spontaneously in most populations, and evolution continues within each clade. We also present evidence that the targets of natural selection change over time, as epistasis and historical contingency alter the strength of selection on different genes. Together, these results show that long-term adaptation to a constant environment can be a more complex and dynamic process than is often assumed.},
keywords = {Demography and Ecology, Genome Evolution, Historical Contingency, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
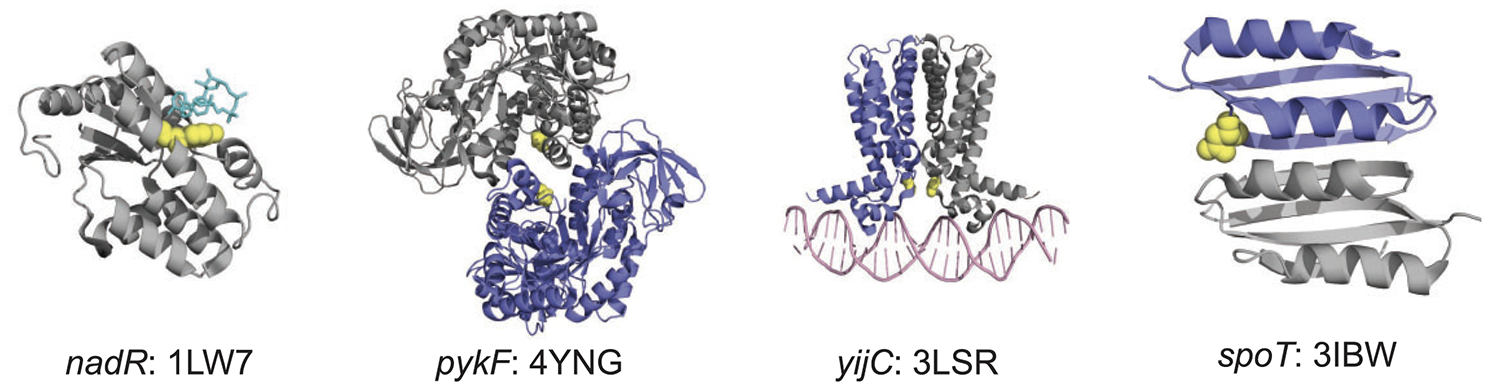
Maddamsetti R; Hatcher P J; Green A G; Williams B L; Marks D S; Lenski R E
Core genes evolve rapidly in the long-term evolution experiment with Escherichia coli Journal Article
Genome Biology and Evolution, vol. 9, no. 4, pp. 1072–1083, 2017, ISSN: 17596653.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Genotypes and Phenotypes, Mutation Rates
@article{Maddamsetti2017,
title = {Core genes evolve rapidly in the long-term evolution experiment with \textit{Escherichia coli}},
author = {Rohan Maddamsetti and Philip J. Hatcher and Anna G. Green and Barry L. Williams and Debora S. Marks and Richard E. Lenski},
url = {https://academic.oup.com/gbe/article/9/4/1072/3100447},
doi = {10.1093/gbe/evx064},
issn = {17596653},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Genome Biology and Evolution},
volume = {9},
number = {4},
pages = {1072--1083},
abstract = {Bacteria can evolve rapidly under positive selection owing to their vast numbers, allowing their genes to diversify by adapting to different environments. We asked whether the same genes that evolve rapidly in the long-term evolution experiment (LTEE) with \textit{Escherichia coli} have also diversified extensively in nature. To make this comparison, we identified ~2000 core genes shared among 60 \textit{E. coli} strains. During the LTEE, core genes accumulated significantly more nonsynonymous mutations than flexible (i.e., noncore) genes. Furthermore, core genes under positive selection in the LTEE are more conserved in nature than the average core gene. In some cases, adaptive mutations appear to modify protein functions, rather than merely knocking them out. The LTEE conditions are novel for \textit{E. coli}, at least in relation to its evolutionary history in nature. The constancy and simplicity of the environment likely favor the complete loss of some unused functions and the fine-tuning of others.},
keywords = {Genome Evolution, Genotypes and Phenotypes, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2016
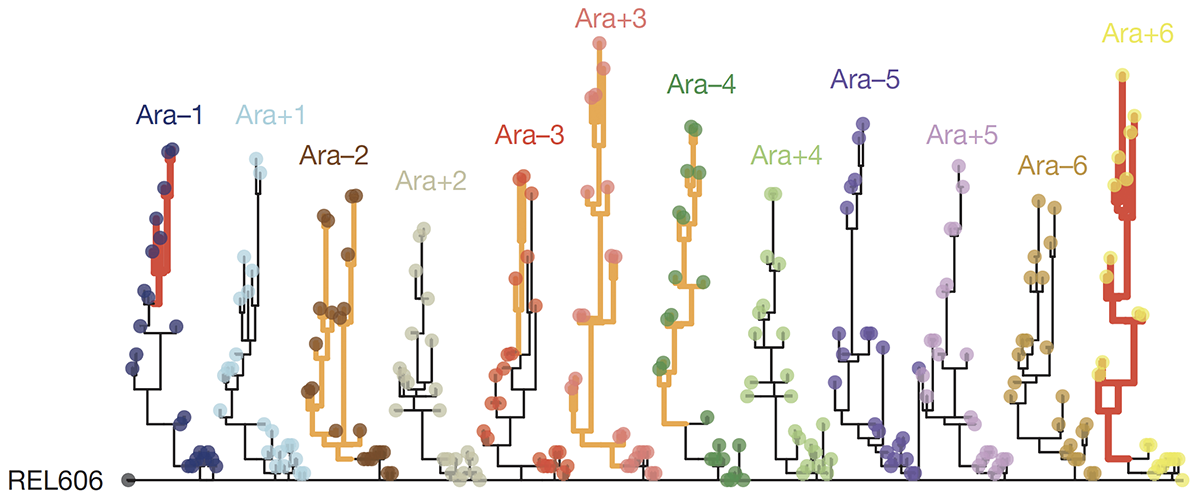
Tenaillon O; Barrick J E; Ribeck N; Deatherage D E; Blanchard J L; Dasgupta A; Wu G C; Wielgoss S; Cruveiller S; Medigue C; Schneider D; Lenski R E
Tempo and mode of genome evolution in a 50,000-generation experiment. Journal Article
Nature, vol. 536, no. 7615, pp. 165–170, 2016, ISSN: 1476-4687.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates, Parallelism and Divergence
@article{Tenaillon2016,
title = {Tempo and mode of genome evolution in a 50,000-generation experiment.},
author = {Olivier Tenaillon and Jeffrey E. Barrick and Noah Ribeck and Daniel E. Deatherage and Jeffrey L. Blanchard and Aurko Dasgupta and Gabriel C. Wu and Sebastien Wielgoss and Stephane Cruveiller and Claudine Medigue and Dominique Schneider and Richard E. Lenski},
url = {http://www.ncbi.nlm.nih.gov/pubmed/27479321},
doi = {10.1038/nature18959},
issn = {1476-4687},
year = {2016},
date = {2016-08-01},
urldate = {2016-08-01},
journal = {Nature},
volume = {536},
number = {7615},
pages = {165--170},
publisher = {Nature Publishing Group},
abstract = {Adaptation by natural selection depends on the rates, effects and interactions of many mutations, making it difficult to determine what proportion of mutations in an evolving lineage are beneficial. Here we analysed 264 complete genomes from 12 \textit{Escherichia coli} populations to characterize their dynamics over 50,000 generations. The populations that retained the ancestral mutation rate support a model in which most fixed mutations are beneficial, the fraction of beneficial mutations declines as fitness rises, and neutral mutations accumulate at a constant rate. We also compared these populations to mutation-accumulation lines evolved under a bottlenecking regime that minimizes selection. Nonsynonymous mutations, intergenic mutations, insertions and deletions are overrepresented in the long-term populations, further supporting the inference that most mutations that reached high frequency were favoured by selection. These results illuminate the shifting balance of forces that govern genome evolution in populations adapting to a new environment.},
keywords = {Genome Evolution, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
2015
Lenski R E; Wiser M J; Ribeck N; Blount Z D; Nahum J R; Morris J J; Zaman L; Turner C B; Wade B D; Maddamsetti R; Burmeister A R; Baird E J; Bundy J; Grant N A; Card K J; Rowles M; Weatherspoon K; Papoulis S E; Sullivan R; Clark C; Mulka J S; Hajela N
Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with Escherichia coli Journal Article
Proceedings of the Royal Society B: Biological Sciences, vol. 282, no. 1821, pp. 20152292, 2015, ISSN: 0962-8452.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Mutation Rates, Parallelism and Divergence
@article{nokey,
title = {Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with \textit{Escherichia coli}},
author = {Richard E. Lenski and Michael J. Wiser and Noah Ribeck and Zachary D. Blount and Joshua R. Nahum and J. Jeffrey Morris and Luis Zaman and Caroline B. Turner and Brian D. Wade and Rohan Maddamsetti and Alita R. Burmeister and Elizabeth J. Baird and Jay Bundy and Nkrumah A. Grant and Kyle J. Card and Maia Rowles and Kiyana Weatherspoon and Spiridon E. Papoulis and Rachel Sullivan and Colleen Clark and Joseph S. Mulka and Neerja Hajela},
url = {https://royalsocietypublishing.org/doi/10.1098/rspb.2015.2292},
doi = {10.1098/rspb.2015.2292},
issn = {0962-8452},
year = {2015},
date = {2015-12-22},
urldate = {2015-12-22},
journal = {Proceedings of the Royal Society B: Biological Sciences},
volume = {282},
number = {1821},
pages = {20152292},
abstract = {Many populations live in environments subject to frequent biotic and abiotic changes. Nonetheless, it is interesting to ask whether an evolving population's mean fitness can increase indefinitely, and potentially without any limit, even in a constant environment. A recent study showed that fitness trajectories of \textit{Escherichia coli} populations over 50 000 generations were better described by a power-law model than by a hyperbolic model. According to the power-law model, the rate of fitness gain declines over time but fitness has no upper limit, whereas the hyperbolic model implies a hard limit. Here, we examine whether the previously estimated power-law model predicts the fitness trajectory for an additional 10 000 generations. To that end, we conducted more than 1100 new competitive fitness assays. Consistent with the previous study, the power-law model fits the new data better than the hyperbolic model. We also analysed the variability in fitness among populations, finding subtle, but significant, heterogeneity in mean fitness. Some, but not all, of this variation reflects differences in mutation rate that evolved over time. Taken together, our results imply that both adaptation and divergence can continue indefinitely—or at least for a long time—even in a constant environment.},
keywords = {Fitness Trajectories, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
2014
Leiby N; Marx C J
PLoS Biology, vol. 12, no. 2, pp. e1001789, 2014, ISSN: 1545-7885.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Mutation Rates
@article{Leiby2014,
title = {Metabolic Erosion Primarily Through Mutation Accumulation, and Not Tradeoffs, Drives Limited Evolution of Substrate Specificity in \textit{Escherichia coli}},
author = {Nicholas Leiby and Christopher J. Marx},
editor = {Nancy A. Moran},
url = {https://dx.plos.org/10.1371/journal.pbio.1001789},
doi = {10.1371/journal.pbio.1001789},
issn = {1545-7885},
year = {2014},
date = {2014-02-01},
urldate = {2014-02-01},
journal = {PLoS Biology},
volume = {12},
number = {2},
pages = {e1001789},
abstract = {Evolutionary adaptation to a constant environment is often accompanied by specialization and a reduction of fitness in other environments. We assayed the ability of the Lenski \textit{Escherichia coli} populations to grow on a range of carbon sources after 50,000 generations of adaptation on glucose. Using direct measurements of growth rates, we demonstrated that declines in performance were much less widespread than suggested by previous results from Biolog assays of cellular respiration. Surprisingly, there were many performance increases on a variety of substrates. In addition to the now famous example of citrate, we observed several other novel gains of function for organic acids that the ancestral strain only marginally utilized. Quantitative growth data also showed that strains with a higher mutation rate exhibited significantly more declines, suggesting that most metabolic erosion was driven by mutation accumulation and not by physiological tradeoffs. These reductions in growth by mutator strains were ameliorated by growth at lower temperature, consistent with the hypothesis that this metabolic erosion is largely caused by destabilizing mutations to the associated enzymes. We further hypothesized that reductions in growth rate would be greatest for substrates used most differently from glucose, and we used flux balance analysis to formulate this question quantitatively. To our surprise, we found no significant relationship between decreases in growth and dissimilarity to glucose metabolism. Taken as a whole, these data suggest that in a single resource environment, specialization does not mainly result as an inevitable consequence of adaptive tradeoffs, but rather due to the gradual accumulation of disabling mutations in unused portions of the genome. },
keywords = {Correlated Responses, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2013
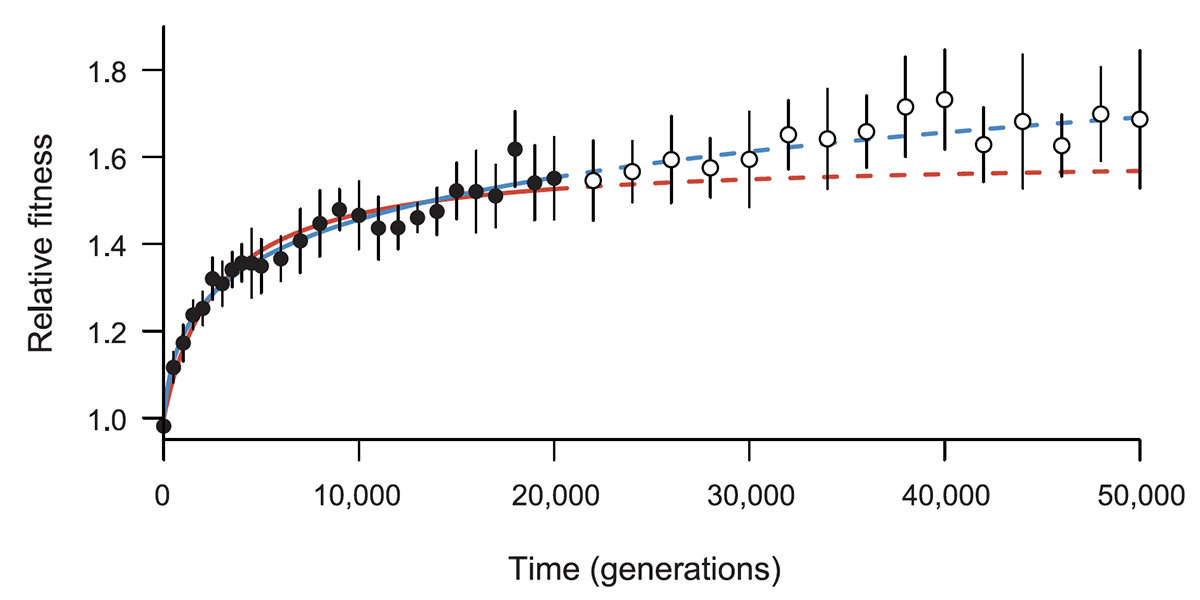
Wiser M J; Ribeck N; Lenski R E
Long-Term Dynamics of Adaptation in Asexual Populations Journal Article
Science, vol. 342, no. 6164, pp. 1364–1367, 2013, ISSN: 0036-8075.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Mutation Rates, Theory and Simulations
@article{Wiser2013,
title = {Long-Term Dynamics of Adaptation in Asexual Populations},
author = {Michael J. Wiser and Noah Ribeck and Richard E. Lenski},
url = {https://www.sciencemag.org/lookup/doi/10.1126/science.1243357},
doi = {10.1126/science.1243357},
issn = {0036-8075},
year = {2013},
date = {2013-12-01},
urldate = {2013-12-01},
journal = {Science},
volume = {342},
number = {6164},
pages = {1364--1367},
abstract = {Experimental studies of evolution have increased greatly in number in recent years, stimulated by the growing power of genomic tools. However, organismal fitness remains the ultimate metric for interpreting these experiments, and the dynamics of fitness remain poorly understood over long time scales. Here, we examine fitness trajectories for 12 \textit{Escherichia coli} populations during 50,000 generations. Mean fitness appears to increase without bound, consistent with a power law. We also derive this power-law relation theoretically by incorporating clonal interference and diminishing-returns epistasis into a dynamical model of changes in mean fitness over time.},
keywords = {Fitness Trajectories, Mutation Rates, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}
Wielgoss S; Barrick J E; Tenaillon O; Wiser M J; Dittmar W J; Cruveiller S; Chane-Woon-Ming B; Médigue C; Lenski R E; Schneider D
Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 110, no. 1, pp. 222–227, 2013, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Theory and Simulations
@article{Wielgoss2013,
title = {Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load},
author = {Sébastien Wielgoss and Jeffrey E. Barrick and Olivier Tenaillon and Michael J. Wiser and W. James Dittmar and Stéphane Cruveiller and Béatrice Chane-Woon-Ming and Claudine Médigue and Richard E. Lenski and Dominique Schneider},
url = {http://www.pnas.org/cgi/doi/10.1073/pnas.1219574110},
doi = {10.1073/pnas.1219574110},
issn = {0027-8424},
year = {2013},
date = {2013-01-01},
urldate = {2013-01-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {110},
number = {1},
pages = {222--227},
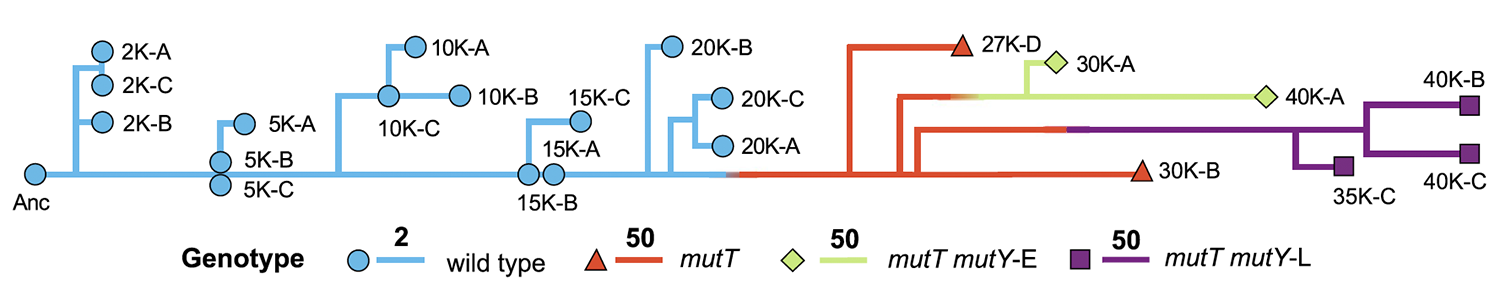
abstract = {Mutations are the ultimate source of heritable variation for evolution. Understanding how mutation rates themselves evolve is thus essential for quantitatively understanding many evolutionary processes. According to theory, mutation rates should be minimized for well-adapted populations living in stable environments, whereas hypermutators may evolve if conditions change. However, the long-term fate of hypermutators is unknown. Using a phylogenomic approach, we found that an adapting \textit{Escherichia coli} population that first evolved a \textit{mutT} hypermutator phenotype was later invaded by two independent lineages with \textit{mutY} mutations that reduced genome-wide mutation rates. Applying neutral theory to synonymous substitutions, we dated the emergence of these mutations and inferred that the \textit{mutT} mutation increased the point-mutation rate by ~150-fold, whereas the \textit{mutY} mutations reduced the rate by ~40-60%, with a corresponding decrease in the genetic load. Thus, the long-term fate of the hypermutators was governed by the selective advantage arising from a reduced mutation rate as the potential for further adaptation declined.},
keywords = {Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}
2012
Blount Z D; Barrick J E; Davidson C J; Lenski R E
Genomic analysis of a key innovation in an experimental Escherichia coli population Journal Article
Nature, vol. 489, no. 7417, pp. 513–518, 2012, ISSN: 0028-0836.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Genome Evolution, Genotypes and Phenotypes, Historical Contingency, Mutation Rates
@article{Blount2012,
title = {Genomic analysis of a key innovation in an experimental \emph{Escherichia coli} population},
author = {Zachary D. Blount and Jeffrey E. Barrick and Carla J. Davidson and Richard E. Lenski},
url = {http://www.nature.com/articles/nature11514},
doi = {10.1038/nature11514},
issn = {0028-0836},
year = {2012},
date = {2012-09-01},
urldate = {2012-09-01},
journal = {Nature},
volume = {489},
number = {7417},
pages = {513--518},
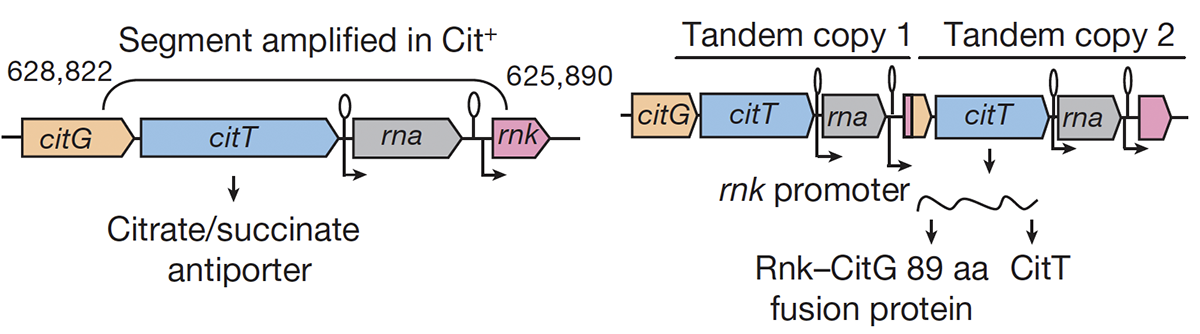
abstract = {Evolutionary novelties have been important in the history of life, but their origins are usually difficult to examine in detail. We previously described the evolution of a novel trait, aerobic citrate utilization (Cit +), in an experimental population of \textit{Escherichia coli}. Here we analyse genome sequences to investigate the history and genetic basis of this trait. At least three distinct clades coexisted for more than 10,000 generations before its emergence. The Cit + trait originated in one clade by a tandem duplication that captured an aerobically expressed promoter for the expression of a previously silent citrate transporter. The clades varied in their propensity to evolve this novel trait, although genotypes able to do so existed in all three clades, implying that multiple potentiating mutations arose during the population's history. Our findings illustrate the importance of promoter capture and altered gene regulation in mediating the exaptation events that often underlie evolutionary innovations.},
keywords = {Citrate Evolution, Genome Evolution, Genotypes and Phenotypes, Historical Contingency, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2011
Wielgoss S; Barrick J E; Tenaillon O; Cruveiller S; Chane-Woon-Ming B; Médigue C; Lenski R E; Schneider D
Mutation Rate Inferred From Synonymous Substitutions in a Long-Term Evolution Experiment With Escherichia coli Journal Article
Genes|Genomes|Genetics, vol. 1, no. 3, pp. 183-186, 2011, ISSN: 2160-1836.
Abstract | Links | BibTeX | Altmetric | Tags: Mutation Rates
@article{nokey,
title = {Mutation Rate Inferred From Synonymous Substitutions in a Long-Term Evolution Experiment With \textit{Escherichia coli}},
author = {Sébastien Wielgoss and Jeffrey E. Barrick and Olivier Tenaillon and Stéphane Cruveiller and Béatrice Chane-Woon-Ming and Claudine Médigue and Richard E. Lenski and Dominique Schneider
},
url = {https://academic.oup.com/g3journal/article/1/3/183/5986556},
doi = {10.1534/g3.111.000406},
issn = {2160-1836},
year = {2011},
date = {2011-08-01},
urldate = {2011-08-01},
journal = {Genes|Genomes|Genetics},
volume = {1},
number = {3},
pages = {183-186},
abstract = {The quantification of spontaneous mutation rates is crucial for a mechanistic understanding of the evolutionary process. In bacteria, traditional estimates using experimental or comparative genetic methods are prone to statistical uncertainty and consequently estimates vary by over one order of magnitude. With the advent of next-generation sequencing, more accurate estimates are now possible. We sequenced 19 \textit{Escherichia coli} genomes from a 40,000-generation evolution experiment and directly inferred the point-mutation rate based on the accumulation of synonymous substitutions. The resulting estimate was 8.9 × 10−11 per base-pair per generation, and there was a significant bias toward increased AT-content. We also compared our results with published genome sequence datasets for other bacterial evolution experiments. Given the power of our approach, our estimate represents the most accurate measure of bacterial base-substitution rates available to date.},
keywords = {Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2009
Barrick J E; Yu D S; Yoon S H; Jeong H; Oh T K; Schneider D; Lenski R E; Kim J F
Genome evolution and adaptation in a long-term experiment with Escherichia coli. Journal Article
Nature, vol. 461, no. 7268, pp. 1243–7, 2009, ISSN: 1476-4687.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Parallelism and Divergence
@article{Barrick2009,
title = {Genome evolution and adaptation in a long-term experiment with \textit{Escherichia coli}.},
author = {Jeffrey E. Barrick and Dong Su Yu and Sung Ho Yoon and Haeyoung Jeong and Tae Kwang Oh and Dominique Schneider and Richard E. Lenski and Jihyun F. Kim},
url = {http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19838166&retmode=ref&cmd=prlinks papers2://publication/doi/10.1038/nature08480 http://www.ncbi.nlm.nih.gov/pubmed/19838166},
doi = {10.1038/nature08480},
issn = {1476-4687},
year = {2009},
date = {2009-10-01},
urldate = {2009-10-01},
journal = {Nature},
volume = {461},
number = {7268},
pages = {1243--7},
publisher = {Nature Publishing Group},
address = {Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, Michigan 48824, USA.},
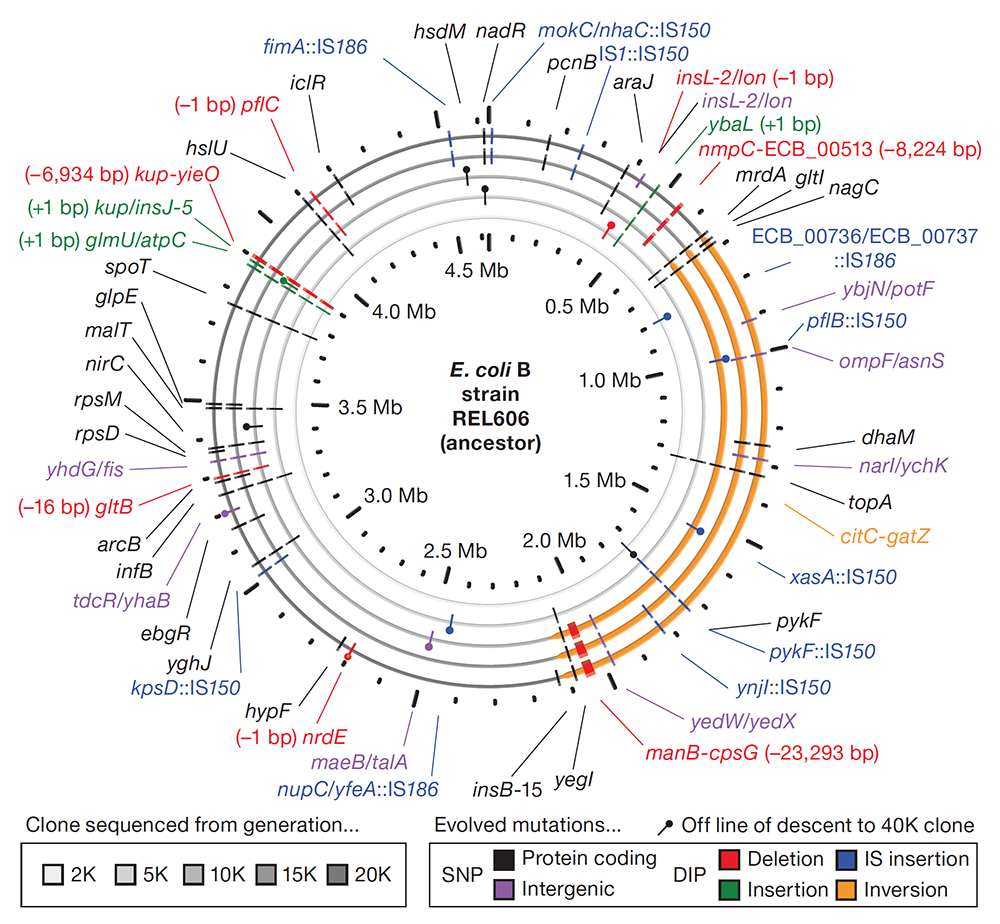
abstract = {The relationship between rates of genomic evolution and organismal adaptation remains uncertain, despite considerable interest. The feasibility of obtaining genome sequences from experimentally evolving populations offers the opportunity to investigate this relationship with new precision. Here we sequence genomes sampled through 40,000 generations from a laboratory population of \textit{Escherichia coli}. Although adaptation decelerated sharply, genomic evolution was nearly constant for 20,000 generations. Such clock-like regularity is usually viewed as the signature of neutral evolution, but several lines of evidence indicate that almost all of these mutations were beneficial. This same population later evolved an elevated mutation rate and accumulated hundreds of additional mutations dominated by a neutral signature. Thus, the coupling between genomic and adaptive evolution is complex and can be counterintuitive even in a constant environment. In particular, beneficial substitutions were surprisingly uniform over time, whereas neutral substitutions were highly variable.},
keywords = {Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
Barrick J E; Lenski R E
Genome-wide Mutational Diversity in an Evolving Population of Escherichia coli Journal Article
Cold Spring Harbor Symposia on Quantitative Biology, vol. 74, pp. 119–129, 2009, ISSN: 0091-7451.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{Barrick2009b,
title = {Genome-wide Mutational Diversity in an Evolving Population of \textit{Escherichia coli}},
author = {Jeffrey E. Barrick and Richard E. Lenski},
url = {http://symposium.cshlp.org/cgi/doi/10.1101/sqb.2009.74.018},
doi = {10.1101/sqb.2009.74.018},
issn = {0091-7451},
year = {2009},
date = {2009-01-01},
urldate = {2009-01-01},
journal = {Cold Spring Harbor Symposia on Quantitative Biology},
volume = {74},
pages = {119--129},
abstract = {The level of genetic variation in a population is the result of a dynamic tension between evolutionary forces. Mutations create variation, certain frequency-dependent interactions may preserve diversity, and natural selection purges variation. New sequencing technologies offer unprecedented opportunities to discover and characterize the diversity present in evolving microbial populations on a whole-genome scale. By sequencing mixed-population samples, we have identified single-nucleotide polymorphisms (SNPs) present at various points in the history of an \textit{Escherichia coli} population that has evolved for almost 20 years from a founding clone. With 50-fold genome coverage, we were able to catch beneficial mutations as they swept to fixation, discover contending beneficial alleles that were eliminated by clonal interference, and detect other minor variants possibly adapted to a new ecological niche. Additionally, there was a dramatic increase in genetic diversity late in the experiment after a mutator phenotype evolved. Still finer-resolution details of the structure of genetic variation and how it changes over time in microbial evolution experiments will enable new applications and quantitative tests of population genetic theory. },
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2003
Lenski R E; Winkworth C L; Riley M A
Rates of DNA Sequence Evolution in Experimental Populations of Escherichia coli During 20,000 Generations Journal Article
Journal of Molecular Evolution, vol. 56, no. 4, pp. 498–508, 2003, ISSN: 0022-2844.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{Lenski2003,
title = {Rates of DNA Sequence Evolution in Experimental Populations of \textit{Escherichia coli} During 20,000 Generations},
author = {Richard E. Lenski and Cynthia L. Winkworth and Margaret A. Riley},
url = {http://link.springer.com/10.1007/s00239-002-2423-0},
doi = {10.1007/s00239-002-2423-0},
issn = {0022-2844},
year = {2003},
date = {2003-04-01},
urldate = {2003-04-01},
journal = {Journal of Molecular Evolution},
volume = {56},
number = {4},
pages = {498--508},
abstract = {We examined rates of DNA sequence evolution in 12 populations of \textit{Escherichia coli} propagated in a glucose minimal medium for 20,000 generations. Previous work saw mutations mediated by mobile elements in these populations, but the extent of other genomic changes was not investigated. Four of the populations evolved defects in DNA repair and became mutators. Some 500 bp was sequenced in each of 36 genes for 50 clones, including 2 ancestral variants, 2 clones from each population at generation 10,000, and 2 from each at generation 20,000. Ten mutations were found in total, all point mutations including mostly synonymous substitutions and nonsynonymous polymorphisms; all 10 were found in mutator populations. We compared the observed sequence evolution to predictions based on different scenarios. The number of synonymous substitutions is lower than predicted from measured mutation rates in \textit{E. coli}, but the number is higher than rates based on comparing \textit{E. coli} and \textit{Salmonella} genomes. Extrapolating to the entire genome, these data predict about 250 synonymous substitutions on average per mutator population, but only about 3 synonymous substitutions per nonmutator population, during 20,000 generations. These data illustrate the challenge of finding sequence variation among bacterial isolates that share such a recent ancestor. However, this limited variation also provides a useful baseline for research aimed at finding the beneficial substitutions in these populations.},
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2000
Cooper V S; Lenski R E
The population genetics of ecological specialization in evolving Escherichia coli populations Journal Article
Nature, vol. 407, no. 6805, pp. 736–739, 2000, ISSN: 0028-0836.
Abstract | Links | BibTeX | Altmetric | Tags: Correlated Responses, Fitness Trajectories, Mutation Rates
@article{Schneider2000b,
title = {The population genetics of ecological specialization in evolving \textit{Escherichia coli} populations},
author = {Vaughn S. Cooper and Richard E. Lenski},
url = {http://www.nature.com/articles/35037572},
doi = {10.1038/35037572},
issn = {0028-0836},
year = {2000},
date = {2000-10-01},
urldate = {2000-10-01},
journal = {Nature},
volume = {407},
number = {6805},
pages = {736--739},
abstract = {When organisms adapt genetically to one environment, they may lose fitness in other environments. Two distinct population genetic processes can produce ecological specialization—mutation accumulation and antagonistic pleiotropy. In mutation accumulation, mutations become fixed by genetic drift in genes that are not maintained by selection; adaptation to one environment and loss of adaptation to another are caused by different mutations. Antagonistic pleiotropy arises from trade-offs, such that the same mutations that are beneficial in one environment are detrimental in another. In general, it is difficult to distinguish between these processes. We analysed the decay of unused catabolic functions in 12 lines of \textit{Escherichia coli} propagated on glucose for 20,000 generations. During that time, several lines evolved high mutation rates. If mutation accumulation is important, their unused functions should decay more than the other lines, but no significant difference was observed. Moreover, most catabolic losses occurred early in the experiment when beneficial mutations were being rapidly fixed, a pattern predicted by antagonistic pleiotropy. Thus, antagonistic pleiotropy appears more important than mutation accumulation for the decay of unused catabolic functions in these populations.},
keywords = {Correlated Responses, Fitness Trajectories, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
1999
Vulic M; Lenski R E; Radman M
Mutation, recombination, and incipient speciation of bacteria in the laboratory Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 96, no. 13, pp. 7348–7351, 1999, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{Vulic1999,
title = {Mutation, recombination, and incipient speciation of bacteria in the laboratory},
author = {M. Vulic and Richard E. Lenski and Miroslav Radman},
url = {http://www.pnas.org/cgi/doi/10.1073/pnas.96.13.7348},
doi = {10.1073/pnas.96.13.7348},
issn = {0027-8424},
year = {1999},
date = {1999-06-01},
urldate = {1999-06-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {96},
number = {13},
pages = {7348--7351},
abstract = {Mutations in the DNA mismatch repair system increase mutation and recombination. They may thereby promote the genetic divergence that underlies speciation, after which the reacquisition of a functional repair system may sustain that divergence by creating a barrier to recombination. We tested several lines of \textit{Escherichia coli}, derived from a common ancestor and evolved for 20,000 generations, for their recombination ability. Some lines, but not others, had become mismatch repair-defective mutators during experimental evolution, providing different opportunities for DNA sequence divergence. We knocked out the repair system in lines that had retained this function, and we restored function to those lines that had become defective. We then estimated recombination rates in various crosses between these repair- deficient and -proficient strains. The effect of the mismatch repair system on recombination was greatest in those lines that had evolved nonfunctional repair, indicating they had undergone more sequence divergence and, consequently, were more sensitive to the recombination-inhibiting effect of a functional repair system. These results demonstrate the establishment of an incipient genetic barrier between formerly identical lines, and they support a model in which the mismatch repair system can influence speciation dynamics through its simultaneous effects on mutation and recombination.},
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
de Visser J A G M; Zeyl C W; Gerrish P J; Blanchard J L; Lenski R E
Diminishing returns from mutation supply rate in asexual populations. Journal Article
Science, vol. 283, pp. 404-406, 1999, ISSN: 00368075.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Mutation Rates
@article{nokey,
title = {Diminishing returns from mutation supply rate in asexual populations.},
author = {J. Arjan G. M. {de Visser} and C. W. Zeyl and Philip J. Gerrish and Jeffrey L. Blanchard and Richard E. Lenski},
url = {https://www.science.org/lookup/doi/10.1126/science.283.5400.404},
doi = {10.1126/science.283.5400.404},
issn = {00368075},
year = {1999},
date = {1999-01-15},
urldate = {1999-01-15},
journal = {Science},
volume = {283},
pages = {404-406},
abstract = {Mutator genotypes with increased mutation rates may be especially important in microbial evolution if genetic adaptation is generally limited by the supply of mutations. In experimental populations of the bacterium \textit{Escherichia coli}, the rate of evolutionary adaptation was proportional to the mutation supply rate only in particular circumstances of small or initially well-adapted populations. These experiments also demonstrate a "speed limit" on adaptive evolution in asexual populations, one that is independent of the mutation supply rate.},
keywords = {Descendant Experiments, Fitness Trajectories, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
Papadopoulos D; Schneider D; Meier-Eiss J; Arber W; Lenski R E; Blot M
Genomic evolution during a 10,000-generation experiment with bacteria Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 96, no. 7, pp. 3807–3812, 1999, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates
@article{Papadopoulos3807,
title = {Genomic evolution during a 10,000-generation experiment with bacteria},
author = {Dimitri Papadopoulos and Dominique Schneider and Jessica Meier-Eiss and Werner Arber and Richard E. Lenski and Michel Blot},
url = {https://www.pnas.org/content/96/7/3807},
doi = {10.1073/pnas.96.7.3807},
issn = {0027-8424},
year = {1999},
date = {1999-01-01},
urldate = {1999-01-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {96},
number = {7},
pages = {3807--3812},
publisher = {National Academy of Sciences},
abstract = {Molecular methods are used widely to measure genetic diversity within populations and determine relationships among species. However, it is difficult to observe genomic evolution in action because these dynamics are too slow in most organisms. To overcome this limitation, we sampled genomes from populations of \textit{Escherichia coli} evolving in the laboratory for 10,000 generations. We analyzed the genomes for restriction fragment length polymorphisms (RFLP) using seven insertion sequences (IS) as probes; most polymorphisms detected by this approach reflect rearrangements (including transpositions) rather than point mutations. The evolving genomes became increasingly different from their ancestor over time. Moreover, tremendous diversity accumulated within each population, such that almost every individual had a different genetic fingerprint after 10,000 generations. As has been often suggested, but not previously shown by experiment, the rates of phenotypic and genomic change were discordant, both across replicate populations and over time within a population. Certain pivotal mutations were shared by all descendants in a population, and these are candidates for beneficial mutations, which are rare and difficult to find. More generally, these data show that the genome is highly dynamic even over a time scale that is, from an evolutionary perspective, very brief. },
keywords = {Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}