2025
Lozzi B; Adepoju L; Espinoza J L; Padgen M; Parra M; Ricco A; Castro-Wallace S; Barrick J E; O’Rourke A
Simulated microgravity triggers a membrane adaptation to stress in E. coli REL606 Journal Article
BMC Microbiology, vol. 25, no. 1, pp. 362, 2025, ISSN: 1471-2180.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genotypes and Phenotypes
@article{Lozzi2025,
title = {Simulated microgravity triggers a membrane adaptation to stress in \textit{E. coli} REL606},
author = {Brittney Lozzi and Lea Adepoju and Josh L. Espinoza and Michael Padgen and Macarena Parra and Antonio Ricco and Sarah Castro-Wallace and Jeffrey E. Barrick and Aubrie O’Rourke},
doi = {10.1186/s12866-025-04064-7},
issn = {1471-2180},
year = {2025},
date = {2025-06-09},
urldate = {2025-06-09},
journal = {BMC Microbiology},
volume = {25},
number = {1},
pages = {362},
abstract = {Investigating the evolution of \textit{Escherichia coli} in microgravity offers valuable insights into microbial adaptation to extreme environments. Here the effects of simulated microgravity (SµG) on gene expression and genome evolution of \textit{E. coli} REL606, a strain evolved terrestrially for 35 years, is explored. The transcriptomic changes for glucose-limited and glucose-replete conditions over 24 h illustrate that SµG increased the expression of genes involved in stress response, biofilm, and metabolism. A greater number of differentially expressed genes related to the general stress response (GSR) and biofilm formation is observed in simulated microgravity cultures under glucose-limited conditions in comparison to glucose-replete conditions. Longer term SµG culture under glucose-limited conditions led to the accumulation of unique mutations when compared to control cultures, particularly in the \textit{mraZ}/\textit{fruR} intergenic region and the \textit{elyC} gene, suggesting changes in peptidoglycan and enterobacterial common antigen (ECA) production. These findings highlight the physiological and genomic adaptations of E. coli to microgravity, offering a foundation for future research into the long-term effects of space conditions on bacterial evolution.},
keywords = {Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
2024
uz-Zaman Md H; D'Alton S; Barrick J E; Ochman H
Promoter capture drives the emergence of proto-genes in Escherichia coli Journal Article
PLoS Biology, vol. 22, pp. e3002418, 2024.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution
@article{uz-Zaman2024,
title = {Promoter capture drives the emergence of proto-genes in \textit{Escherichia coli}},
author = {Md. Hassan uz-Zaman and Simon D'Alton and Jeffrey E. Barrick and Howard Ochman},
doi = {10.1371/journal.pbio.3002418},
year = {2024},
date = {2024-05-07},
urldate = {2023-11-17},
journal = {PLoS Biology},
volume = {22},
pages = {e3002418},
publisher = {Cold Spring Harbor Laboratory},
abstract = {The phenomenon of de novo gene birth-the emergence of genes from non-genic sequences-has received considerable attention due to the widespread occurrence of genes that are unique to particular species or genomes. Most instances of de novo gene birth have been recognized through comparative analyses of genome sequences in eukaryotes, despite the abundance of novel, lineage-specific genes in bacteria and the relative ease with which bacteria can be studied in an experimental context. Here, we explore the genetic record of the \textit{Escherichia coli} long-term evolution experiment (LTEE) for changes indicative of "proto-genic" phases of new gene birth in which non-genic sequences evolve stable transcription and/or translation. Over the time span of the LTEE, non-genic regions are frequently transcribed, translated and differentially expressed, with levels of transcription across low-expressed regions increasing in later generations of the experiment. Proto-genes formed downstream of new mutations result either from insertion element activity or chromosomal translocations that fused preexisting regulatory sequences to regions that were not expressed in the LTEE ancestor. Additionally, we identified instances of proto-gene emergence in which a previously unexpressed sequence was transcribed after formation of an upstream promoter, although such cases were rare compared to those caused by recruitment of preexisting promoters. Tracing the origin of the causative mutations, we discovered that most occurred early in the history of the LTEE, often within the first 20,000 generations, and became fixed soon after emergence. Our findings show that proto-genes emerge frequently within evolving populations, can persist stably, and can serve as potential substrates for new gene formation. },
keywords = {Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
2023
Barrick J E; Blount Z D; Lake D M; Dwenger J H; Chavarria-Palma J E; Izutsu M; Wiser M J
Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli Journal Article
Journal of Visualized Experiments, vol. 198, pp. e65342, 2023, ISSN: 1940-087X.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Methods and Miscellaneous
@article{Barrick2023,
title = {Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli},
author = {Jeffrey E. Barrick and Zachary D. Blount and Devin M. Lake and Jack H. Dwenger and Jesus E. Chavarria-Palma and Minako Izutsu and Michael J. Wiser},
doi = {10.3791/65342},
issn = {1940-087X},
year = {2023},
date = {2023-08-18},
urldate = {2023-08-18},
journal = {Journal of Visualized Experiments},
volume = {198},
pages = {e65342},
abstract = {The Long-Term Evolution Experiment (LTEE) has followed twelve populations of \textit{Escherichia coli} as they have adapted to a simple laboratory environment for more than 35 years and 77,000 bacterial generations. The setup and procedures used in the LTEE epitomize reliable and reproducible methods for studying microbial evolution. In this protocol, we first describe how the LTEE populations are transferred to fresh medium and cultured each day. Then, we describe how the LTEE populations are regularly checked for possible signs of contamination and archived to provide a permanent frozen "fossil record" for later study. Multiple safeguards included in these procedures are designed to prevent contamination, detect various problems when they occur, and recover from disruptions without appreciably setting back the progress of the experiment. One way that the overall tempo and character of evolutionary changes are monitored in the LTEE is by measuring the competitive fitness of populations and strains from the experiment. We describe how co-culture competition assays are conducted and provide both a spreadsheet and an R package (fitnessR) for calculating relative fitness from the results. Over the course of the LTEE, the behaviors of some populations have changed in interesting ways, and new technologies like whole-genome sequencing have provided additional avenues for investigating how the populations have evolved. We end by discussing how the original LTEE procedures have been updated to accommodate or take advantage of these changes. This protocol will be useful for researchers who use the LTEE as a model system for studying connections between evolution and genetics, molecular biology, systems biology, and ecology. More broadly, the LTEE provides a tried-and-true template for those who are beginning their own evolution experiments with new microbes, environments, and questions. },
keywords = {Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2021
Consuegra J; Gaffé J; Lenski R E; Hindré T; Barrick J E; Tenaillon O; Schneider D
Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria Journal Article
Nature Communications, vol. 12, no. 1, pp. 980, 2021, ISSN: 2041-1723.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Mutation Rates
@article{Consuegra2021,
title = {Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria},
author = {Jessika Consuegra and Joël Gaffé and Richard E. Lenski and Thomas Hindré and Jeffrey E. Barrick and Olivier Tenaillon and Dominique Schneider},
url = {http://www.nature.com/articles/s41467-021-21210-7},
doi = {https://doi.org/10.1038/s41467-021-21210-7},
issn = {2041-1723},
year = {2021},
date = {2021-12-01},
urldate = {2021-12-01},
journal = {Nature Communications},
volume = {12},
number = {1},
pages = {980},
publisher = {Springer US},
abstract = {Insertion sequences (IS) are ubiquitous bacterial mobile genetic elements, and the mutations they cause can be deleterious, neutral, or beneficial. The long-term dynamics of IS elements and their effects on bacteria are poorly understood, including whether they are primarily genomic parasites or important drivers of adaptation by natural selection. Here, we investigate the dynamics of IS elements and their contribution to genomic evolution and fitness during a long-term experiment with \textit{Escherichia coli}. IS elements account for ~35% of the mutations that reached high frequency through 50,000 generations in those populations that retained the ancestral point-mutation rate. In mutator populations, IS-mediated mutations are only half as frequent in absolute numbers. In one population, an exceptionally high ~8-fold increase in IS 150 copy number is associated with the beneficial effects of early insertion mutations; however, this expansion later slowed down owing to reduced IS 150 activity. This population also achieves the lowest fitness, suggesting that some avenues for further adaptation are precluded by the IS 150 -mediated mutations. More generally, across all populations, we find that higher IS activity becomes detrimental to adaptation over evolutionary time. Therefore, IS-mediated mutations can both promote and constrain evolvability.},
keywords = {Fitness Trajectories, Genome Evolution, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
Deatherage D E; Barrick J E
High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing Journal Article
Cell Systems, vol. 12, no. 12, pp. 1187-1200, 2021, ISSN: 24054712.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous
@article{DEATHERAGE2021,
title = {High-throughput characterization of mutations in genes that drive clonal evolution using multiplex adaptome capture sequencing},
author = {Daniel E. Deatherage and Jeffrey E. Barrick},
url = {https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8678185/},
doi = {10.1016/j.cels.2021.08.011},
issn = {24054712},
year = {2021},
date = {2021-09-01},
urldate = {2021-09-01},
journal = {Cell Systems},
volume = {12},
number = {12},
pages = {1187-1200},
abstract = {Understanding how cells are likely to evolve can guide medical interventions and bioengineering efforts that must contend with unwanted mutations. The adaptome of a cell—the neighborhood of genetic changes that are most likely to drive adaptation in a given environment—can be mapped by tracking rare beneficial variants during the early stages of clonal evolution. We used multiplex adaptome capture sequencing (mAdCap-seq), a procedure that combines unique molecular identifiers and hybridization-based enrichment, to characterize mutations in eight \textit{Escherichia coli} genes known to be under selection in a laboratory environment. We tracked 301 mutations at frequencies as low as 0.01% and inferred the fitness effects of 240 of these mutations. There were distinct molecular signatures of selection on protein structure and function for the three genes with the most beneficial mutations. Our results demonstrate how mAdCap-seq can be used to deeply profile a targeted portion of a cell's adaptome.},
keywords = {Descendant Experiments, Fitness Trajectories, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
Card K J; Thomas M D; Graves J L; Barrick J E; Lenski R E
Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with Escherichia coli Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 118, no. 5, pp. e2016886118, 2021, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Genome Evolution, Historical Contingency
@article{Card2021,
title = {Genomic evolution of antibiotic resistance is contingent on genetic background following a long-term experiment with \textit{Escherichia coli}},
author = {Kyle J. Card and Misty D. Thomas and Joseph L. Graves and Jeffrey E. Barrick and Richard E. Lenski},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.2016886118},
doi = {10.1073/pnas.2016886118},
issn = {0027-8424},
year = {2021},
date = {2021-02-01},
urldate = {2021-02-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {118},
number = {5},
pages = {e2016886118},
abstract = {Antibiotic resistance is a growing health concern. Efforts to control resistance would benefit from an improved ability to forecast when and how it will evolve. Epistatic interactions between mutations can promote divergent evolutionary trajectories, which complicates our ability to predict evolution. We recently showed that differences between genetic backgrounds can lead to idiosyncratic responses in the evolvability of phenotypic resistance, even among closely related \textit{Escherichia coli} strains. In this study, we examined whether a strain's genetic background also influences the genotypic evolution of resistance. Do lineages founded by different genotypes take parallel or divergent mutational paths to achieve their evolved resistance states? We addressed this question by sequencing the complete genomes of antibiotic-resistant clones that evolved from several different genetic starting points during our earlier experiments. We first validated our statistical approach by quantifying the specificity of genomic evolution with respect to antibiotic treatment. As expected, mutations in particular genes were strongly associated with each drug. Then, we determined that replicate lines evolved from the same founding genotypes had more parallel mutations at the gene level than lines evolved from different founding genotypes, although these effects were more subtle than those showing antibiotic specificity. Taken together with our previous work, we conclude that historical contingency can alter both genotypic and phenotypic pathways to antibiotic resistance.},
keywords = {Descendant Experiments, Genome Evolution, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
Gifford I; Dasgupta A; Barrick J E
Rates of gene conversions between Escherichia coli ribosomal operons Journal Article
G3: Genes, Genomes, Genetics, vol. 11, no. 2, pp. jkaa002, 2021, ISSN: 21601836.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments, Mutation Rates
@article{Gifford2021,
title = {Rates of gene conversions between \emph{Escherichia coli} ribosomal operons},
author = {Isaac Gifford and Aurko Dasgupta and Jeffrey E. Barrick},
url = {https://academic.oup.com/g3journal/article/11/2/jkaa002/5974039},
doi = {10.1093/g3journal/jkaa002},
issn = {21601836},
year = {2021},
date = {2021-01-01},
urldate = {2021-01-01},
journal = {G3: Genes, Genomes, Genetics},
volume = {11},
number = {2},
pages = {jkaa002},
abstract = {Due to their universal presence and high sequence conservation, ribosomal RNA (rRNA) sequences are used widely in phylogenetics for inferring evolutionary relationships between microbes and in metagenomics for analyzing the composition of microbial communities. Most microbial genomes encode multiple copies of rRNA genes to supply cells with sufficient capacity for protein synthesis. These copies typically undergo concerted evolution that keeps their sequences identical, or nearly so, due to gene conversion, a type of intragenomic recombination that changes one copy of a homologous sequence to exactly match another. Widely varying rates of rRNA gene conversion have previously been estimated by comparative genomics methods and using genetic reporter assays. To more directly measure rates of rRNA intragenomic recombination, we sequenced the seven \textit{Escherichia coli} rRNA operons in 15 lineages that were evolved for ~13,750 generations with frequent single-cell bottlenecks that reduce the effects of selection. We identified 38 gene conversion events and estimated an overall rate of intragenomic recombination within the 16S and 23S genes between rRNA copies of 3.6 × 10^{−4} per genome per generation or 8.6 × 10^{6} per rRNA operon per homologous donor operon per generation. This rate varied only slightly from random expectations at different sites within the rRNA genes and between rRNA operons located at different positions in the genome. Our accurate estimate of the rate of rRNA gene conversions fills a gap in our quantitative understanding of how ribosomal sequences and other multicopy elements diversify and homogenize during microbial genome evolution.},
keywords = {Descendant Experiments, Mutation Rates},
pubstate = {published},
tppubtype = {article}
}
2020
Blount Z D; Maddamsetti R; Grant N A; Ahmed S T; Jagdish T; Baxter J A; Sommerfeld B A; Tillman A; Moore J; Slonczewski J L; Barrick J E; Lenski R E
Genomic and phenotypic evolution of Escherichia coli in a novel citrate-only resource environment Journal Article
eLife, vol. 9, pp. 1–64, 2020, ISSN: 2050-084X.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes
@article{Blount2020,
title = {Genomic and phenotypic evolution of \textit{Escherichia coli} in a novel citrate-only resource environment},
author = {Zachary D. Blount and Rohan Maddamsetti and Nkrumah A. Grant and Sumaya T. Ahmed and Tanush Jagdish and Jessica A. Baxter and Brooke A. Sommerfeld and Alice Tillman and Jeremy Moore and Joan L. Slonczewski and Jeffrey E. Barrick and Richard E. Lenski},
url = {https://elifesciences.org/articles/55414},
doi = {10.7554/eLife.55414},
issn = {2050-084X},
year = {2020},
date = {2020-05-01},
urldate = {2020-05-01},
journal = {eLife},
volume = {9},
pages = {1--64},
abstract = {Evolutionary innovations allow populations to colonize new ecological niches. We previously reported that aerobic growth on citrate (Cit^{+}) evolved in an \textit{Escherichia coli} population during adaptation to a minimal glucose medium containing citrate (DM25). Cit^{+} variants can also grow in citrate-only medium (DM0), a novel environment for \textit{E. coli}. To study adaptation to this niche, we founded two sets of Cit^{+} populations and evolved them for 2500 generations in DM0 or DM25. The evolved lineages acquired numerous parallel mutations, many mediated by transposable elements. Several also evolved amplifications of regions containing the \textit{maeA} gene. Unexpectedly, some evolved populations and clones show apparent declines in fitness. We also found evidence of substantial cell death in Cit^{+} clones. Our results thus demonstrate rapid trait refinement and adaptation to the new citrate niche, while also suggesting a recalcitrant mismatch between \textit{E. coli} physiology and growth on citrate.},
keywords = {Citrate Evolution, Demography and Ecology, Descendant Experiments, Genotypes and Phenotypes},
pubstate = {published},
tppubtype = {article}
}
Barrick J E; Deatherage D E; Lenski R E
Banzhaf, Wolfgang; Cheng, Betty H C; Deb, Kalyanmoy; Holekamp, Kay E; Lenski, Richard E; Ofria, Charles; Pennock, Robert T; Punch, William F; Whittaker, Danielle J (Ed.): Evolution in Action: Past, Present and Future: A Festschrift in Honor of Erik D. Goodman, pp. 77–89, Springer International Publishing, Cham, 2020, ISBN: 978-3-030-39831-6.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories
@inbook{Barrick2020,
title = {A Test of the Repeatability of Measurements of Relative Fitness in the Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Jeffrey E. Barrick and Daniel E. Deatherage and Richard E. Lenski},
editor = {Wolfgang Banzhaf and Betty H C Cheng and Kalyanmoy Deb and Kay E Holekamp and Richard E Lenski and Charles Ofria and Robert T Pennock and William F Punch and Danielle J Whittaker},
url = {http://link.springer.com/10.1007/978-3-030-39831-6_8},
doi = {10.1007/978-3-030-39831-6_8},
isbn = {978-3-030-39831-6},
year = {2020},
date = {2020-01-01},
urldate = {2020-01-01},
booktitle = {Evolution in Action: Past, Present and Future: A Festschrift in Honor of Erik D. Goodman},
pages = {77--89},
publisher = {Springer International Publishing},
address = {Cham},
abstract = {Experimental studies of evolution using microbes have a long tradition, and these studies have increased greatly in number and scope in recent decades. Most such experiments have been short in duration, typically running for weeks or months. A venerable exception, the long-term evolution experiment (LTEE) with \textit{Escherichia coli} has continued for 30 years and 70,000 bacterial generations. The LTEE has become one of the cornerstones of the field of experimental evolution, in general, and the BEACON Center for the Study of Evolution in Action, in particular. Science laboratories and experiments usually have finite lifespans, but we hope that the LTEE can continue far into the future. There are practical issues associated with maintaining such a long-term experiment. One issue, which we address here, is whether key measurements made at one time and place are reproducible, within reasonable limits, at other times and places. This issue comes to the forefront when one considers moving an experiment like the LTEE from one lab to another. To that end, the Barrick lab at The University of Texas at Austin, measured the fitness values of samples from the 12 LTEE populations at 2,000, 10,000, and 50,000 generations and compared the new data to data previously obtained at Michigan State University. On balance, the datasets agree very well. More generally, this finding shows the value of simplicity in experimental design, such as using a chemically defined growth medium and appropriately storing samples from microbiological experiments. Even so, one must be vigilant in checking assumptions and procedures given the potential for uncontrolled factors (e.g., water quality) to affect outcomes. This vigilance is perhaps especially important for a trait like fitness, which integrates all aspects of organismal performance and may therefore be sensitive to any number of subtle environmental influences.},
keywords = {Fitness Trajectories},
pubstate = {published},
tppubtype = {inbook}
}
2018
Leon D; D'Alton S; Quandt E M; Barrick J E
Innovation in an E. coli evolution experiment is contingent on maintaining adaptive potential until competition subsides Journal Article
PLOS Genetics, vol. 14, no. 4, pp. e1007348, 2018, ISSN: 1553-7404.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Demography and Ecology, Historical Contingency
@article{nokey,
title = {Innovation in an \textit{E. coli} evolution experiment is contingent on maintaining adaptive potential until competition subsides},
author = {Dacia Leon and Simon D'Alton and Erik M. Quandt and Jeffrey E. Barrick},
url = {https://dx.plos.org/10.1371/journal.pgen.1007348},
doi = {10.1371/journal.pgen.1007348},
issn = {1553-7404},
year = {2018},
date = {2018-04-12},
urldate = {2018-04-12},
journal = {PLOS Genetics},
volume = {14},
number = {4},
pages = {e1007348},
abstract = {Key innovations are disruptive evolutionary events that enable a species to escape constraints and rapidly diversify. After 15 years of the Lenski long-term evolution experiment with \textit{Escherichia coli}, cells in one of the twelve populations evolved the ability to utilize citrate, an abundant but previously untapped carbon source in the environment. Descendants of these cells became dominant in the population and subsequently diversified as a consequence of invading this vacant niche. Mutations responsible for the appearance of rudimentary citrate utilization and for refining this ability have been characterized. However, the complete nature of the genetic and/or ecological events that set the stage for this key innovation is unknown. In particular, it is unclear why it took so long for citrate utilization to evolve and why it still has evolved in only one of the twelve \textit{E. coli} populations after 30 years of the Lenski experiment. In this study, we recapitulated the initial mutation needed to evolve citrate utilization in strains isolated from throughout the first 31,500 generations of the history of this population. We found that there was already a slight fitness benefit for this mutation in the original ancestor of the evolution experiment and in other early isolates. However, evolution of citrate utilization was blocked at this point due to competition with other mutations that improved fitness in the original niche. Subsequently, an anti-potentiated genetic background evolved in which it was deleterious to evolve rudimentary citrate utilization. Only later, after further mutations accumulated that restored the benefit of this first-step mutation and the overall rate of adaptation in the population slowed, was citrate utilization likely to evolve. Thus, intense competition and the types of mutations that it favors can lead to short-sighted evolutionary trajectories that hide a stepping stone needed to access a key innovation from many future generations.},
keywords = {Citrate Evolution, Demography and Ecology, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
2017
Deatherage D E; Kepner J L; Bennett A F; Lenski R E; Barrick J E
Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 114, no. 10, pp. E1904–E1912, 2017, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Descendant Experiments
@article{Deatherage2017,
title = {Specificity of genome evolution in experimental populations of \textit{Escherichia coli} evolved at different temperatures},
author = {Daniel E. Deatherage and Jamie L. Kepner and Albert F. Bennett and Richard E. Lenski and Jeffrey E. Barrick},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.1616132114},
doi = {10.1073/pnas.1616132114},
issn = {0027-8424},
year = {2017},
date = {2017-03-01},
urldate = {2017-03-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {114},
number = {10},
pages = {E1904--E1912},
abstract = {Isolated populations derived from a common ancestor are expected to diverge genetically and phenotypically as they adapt to different local environments. To examine this process, 30 populations of \textit{Escherichia coli} were evolved for 2,000 generations, with six in each of five different thermal regimes: constant 20 °C, 32 °C, 37 °C, 42 °C, and daily alternations between 32 °C and 42 °C. Here, we sequenced the genomes of one endpoint clone from each population to test whether the history of adaptation in different thermal regimes was evident at the genomic level. The evolved strains had accumulated ∼5.3 mutations, on average, and exhibited distinct signatures of adaptation to the different environments. On average, two strains that evolved under the same regime exhibited ∼17% overlap in which genes were mutated, whereas pairs that evolved under different conditions shared only ∼4%. For example, all six strains evolved at 32 °C had mutations in nadR , whereas none of the other 24 strains did. However, a population evolved at 37 °C for an additional 18,000 generations eventually accumulated mutations in the signature genes strongly associated with adaptation to the other temperature regimes. Two mutations that arose in one temperature treatment tended to be beneficial when tested in the others, although less so than in the regime in which they evolved. These findings demonstrate that genomic signatures of adaptation can be highly specific, even with respect to subtle environmental differences, but that this imprint may become obscured over longer timescales as populations continue to change and adapt to the shared features of their environments.},
keywords = {Descendant Experiments},
pubstate = {published},
tppubtype = {article}
}
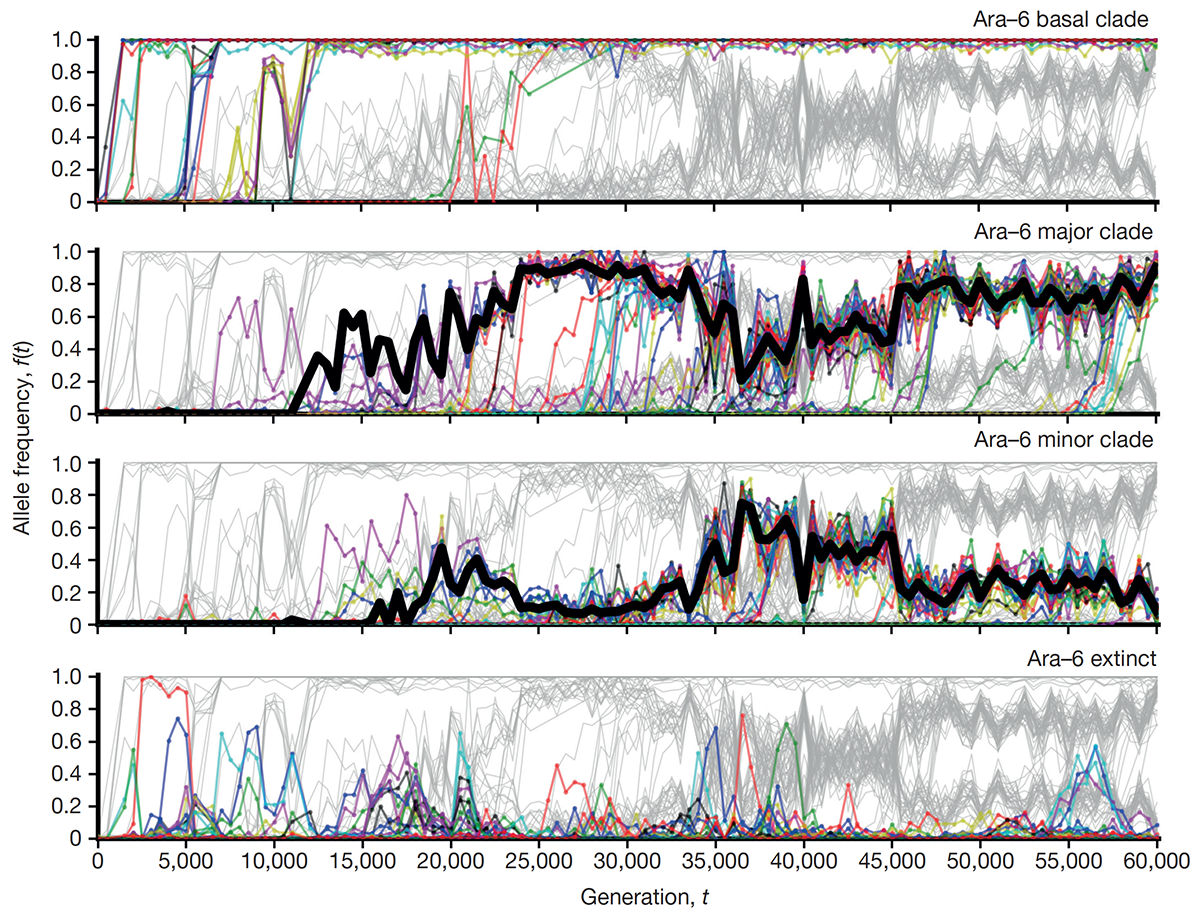
Good B H; McDonald M J; Barrick J E; Lenski R E; Desai M M
The dynamics of molecular evolution over 60,000 generations Journal Article
Nature, vol. 551, no. 7678, pp. 45–50, 2017, ISSN: 14764687.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Genome Evolution, Historical Contingency, Mutation Rates, Parallelism and Divergence
@article{Good2017,
title = {The dynamics of molecular evolution over 60,000 generations},
author = {Benjamin H. Good and Michael J. McDonald and Jeffrey E. Barrick and Richard E. Lenski and Michael M. Desai},
url = {http://dx.doi.org/10.1038/nature24287},
doi = {10.1038/nature24287},
issn = {14764687},
year = {2017},
date = {2017-01-01},
urldate = {2017-01-01},
journal = {Nature},
volume = {551},
number = {7678},
pages = {45--50},
publisher = {Nature Publishing Group},
abstract = {The outcomes of evolution are determined by a stochastic dynamical process that governs how mutations arise and spread through a population. However, it is difficult to observe these dynamics directly over long periods and across entire genomes. Here we analyse the dynamics of molecular evolution in twelve experimental populations of \textit{Escherichia coli}, using whole-genome metagenomic sequencing at five hundred-generation intervals through sixty thousand generations. Although the rate of fitness gain declines over time, molecular evolution is characterized by signatures of rapid adaptation throughout the duration of the experiment, with multiple beneficial variants simultaneously competing for dominance in each population. Interactions between ecological and evolutionary processes play an important role, as long-term quasi-stable coexistence arises spontaneously in most populations, and evolution continues within each clade. We also present evidence that the targets of natural selection change over time, as epistasis and historical contingency alter the strength of selection on different genes. Together, these results show that long-term adaptation to a constant environment can be a more complex and dynamic process than is often assumed.},
keywords = {Demography and Ecology, Genome Evolution, Historical Contingency, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
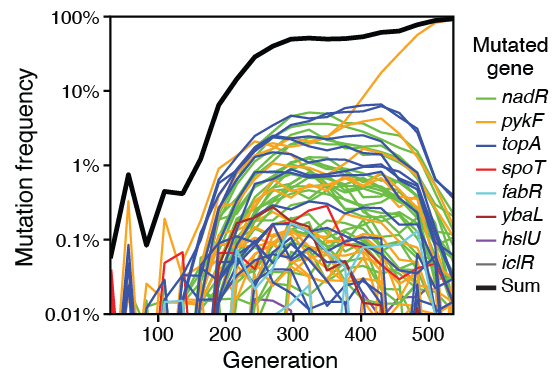
2016
Tenaillon O; Barrick J E; Ribeck N; Deatherage D E; Blanchard J L; Dasgupta A; Wu G C; Wielgoss S; Cruveiller S; Medigue C; Schneider D; Lenski R E
Tempo and mode of genome evolution in a 50,000-generation experiment. Journal Article
Nature, vol. 536, no. 7615, pp. 165–170, 2016, ISSN: 1476-4687.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution, Mutation Rates, Parallelism and Divergence
@article{Tenaillon2016,
title = {Tempo and mode of genome evolution in a 50,000-generation experiment.},
author = {Olivier Tenaillon and Jeffrey E. Barrick and Noah Ribeck and Daniel E. Deatherage and Jeffrey L. Blanchard and Aurko Dasgupta and Gabriel C. Wu and Sebastien Wielgoss and Stephane Cruveiller and Claudine Medigue and Dominique Schneider and Richard E. Lenski},
url = {http://www.ncbi.nlm.nih.gov/pubmed/27479321},
doi = {10.1038/nature18959},
issn = {1476-4687},
year = {2016},
date = {2016-08-01},
urldate = {2016-08-01},
journal = {Nature},
volume = {536},
number = {7615},
pages = {165--170},
publisher = {Nature Publishing Group},
abstract = {Adaptation by natural selection depends on the rates, effects and interactions of many mutations, making it difficult to determine what proportion of mutations in an evolving lineage are beneficial. Here we analysed 264 complete genomes from 12 \textit{Escherichia coli} populations to characterize their dynamics over 50,000 generations. The populations that retained the ancestral mutation rate support a model in which most fixed mutations are beneficial, the fraction of beneficial mutations declines as fitness rises, and neutral mutations accumulate at a constant rate. We also compared these populations to mutation-accumulation lines evolved under a bottlenecking regime that minimizes selection. Nonsynonymous mutations, intergenic mutations, insertions and deletions are overrepresented in the long-term populations, further supporting the inference that most mutations that reached high frequency were favoured by selection. These results illuminate the shifting balance of forces that govern genome evolution in populations adapting to a new environment.},
keywords = {Genome Evolution, Mutation Rates, Parallelism and Divergence},
pubstate = {published},
tppubtype = {article}
}
2015
Maddamsetti R; Hatcher P J; Cruveiller S; Medigue C; Barrick J E; Lenski R E
Molecular Biology and Evolution, vol. 32, no. 11, 2015, ISSN: 0737-4038.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution
@article{Maddamsetti2015,
title = {Synonymous Genetic Variation in Natural Isolates of \textit{Escherichia coli} Does Not Predict Where Synonymous Substitutions Occur in a Long-Term Experiment},
author = {Rohan Maddamsetti and Philip J. Hatcher and Stephane Cruveiller and Claudine Medigue and Jeffrey E. Barrick and Richard E. Lenski},
url = {https://academic.oup.com/mbe/article-lookup/doi/10.1093/molbev/msv161},
doi = {10.1093/molbev/msv161},
issn = {0737-4038},
year = {2015},
date = {2015-11-01},
urldate = {2015-11-01},
journal = {Molecular Biology and Evolution},
volume = {32},
number = {11},
abstract = {Synonymous genetic differences vary by more than 20-fold among genes in natural isolates of \textit{Escherichia coli}. One hypothesis to explain this heterogeneity is that genes with high levels of synonymous variation mutate at higher rates than genes with low synonymous variation. If so, then one would expect to observe similar mutational patterns in evolution experiments. In fact, however, the pattern of synonymous substitutions in a long-term evolution experiment with \textit{E. coli} does not support this hypothesis. In particular, the extent of synonymous variation across genes in that experiment does not reflect the variation observed in natural isolates of \textit{E. coli}. Instead, gene length alone predicts with high accuracy the prevalence of synonymous changes in the experimental populations. We hypothesize that patterns of synonymous variation in natural \textit{E. coli} populations are instead caused by differences across genomic regions in their effective population size that, in turn, reflect different histories of recombination, horizontal gene transfer, selection, and population structure.},
keywords = {Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
Quandt E M; Gollihar J; Blount Z D; Ellington A D; Georgiou G; Barrick J E
Fine-tuning citrate synthase flux potentiates and refines metabolic innovation in the Lenski evolution experiment. Journal Article
eLife, vol. 4, no. October, pp. e09696, 2015, ISSN: 2050-084X.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Genotypes and Phenotypes, Historical Contingency
@article{Quandt2015,
title = {Fine-tuning citrate synthase flux potentiates and refines metabolic innovation in the Lenski evolution experiment.},
author = {Erik M. Quandt and Jimmy Gollihar and Zachary D. Blount and Andrew D. Ellington and George Georgiou and Jeffrey E. Barrick},
url = {http://www.ncbi.nlm.nih.gov/pubmed/26465114
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4718724},
doi = {10.7554/eLife.09696},
issn = {2050-084X},
year = {2015},
date = {2015-10-01},
urldate = {2015-10-01},
journal = {eLife},
volume = {4},
number = {October},
pages = {e09696},
abstract = {Evolutionary innovations that enable organisms to colonize new ecological niches are rare compared to gradual evolutionary changes in existing traits. We discovered that key mutations in the \textit{gltA} gene, which encodes citrate synthase (CS), occurred both before and after \textit{Escherichia coli} gained the ability to grow aerobically on citrate (Cit(+) phenotype) during the Lenski long-term evolution experiment. The first \textit{gltA} mutation, which increases CS activity by disrupting NADH-inhibition of this enzyme, is beneficial for growth on the acetate and contributed to preserving the rudimentary Cit(+) trait from extinction when it first evolved. However, after Cit(+) was refined by further mutations, this potentiating \textit{gltA} mutation became deleterious to fitness. A second wave of beneficial \textit{gltA} mutations then evolved that reduced CS activity to below the ancestral level. Thus, dynamic reorganization of central metabolism made colonizing this new nutrient niche contingent on both co-opting and overcoming a history of prior adaptation.},
keywords = {Citrate Evolution, Genotypes and Phenotypes, Historical Contingency},
pubstate = {published},
tppubtype = {article}
}
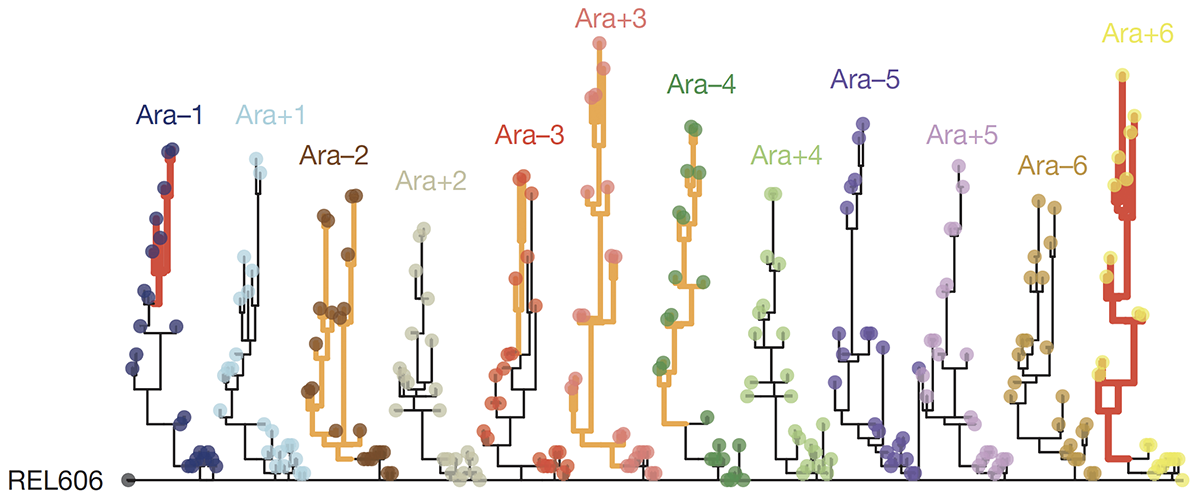
Maddamsetti R; Lenski R E; Barrick J E
Adaptation, Clonal Interference, and Frequency-Dependent Interactions in a Long-Term Evolution Experiment with Escherichia coli Journal Article
Genetics, vol. 200, no. 2, pp. 619-631, 2015, ISSN: 1943-2631.
Abstract | Links | BibTeX | Altmetric | Tags: Demography and Ecology, Fitness Trajectories, Genome Evolution
@article{nokey,
title = {Adaptation, Clonal Interference, and Frequency-Dependent Interactions in a Long-Term Evolution Experiment with \emph{Escherichia coli}},
author = {Rohan Maddamsetti and Richard E. Lenski and Jeffrey E. Barrick},
url = {https://academic.oup.com/genetics/article/200/2/619/5936186},
doi = {10.1534/genetics.115.176677},
issn = {1943-2631},
year = {2015},
date = {2015-04-24},
urldate = {2015-04-24},
journal = {Genetics},
volume = {200},
number = {2},
pages = {619-631},
abstract = {Twelve replicate populations of \textit{Escherichia coli} have been evolving in the laboratory for >25 years and 60,000 generations. We analyzed bacteria from whole-population samples frozen every 500 generations through 20,000 generations for one well-studied population, called Ara−1. By tracking 42 known mutations in these samples, we reconstructed the history of this population’s genotypic evolution over this period. The evolutionary dynamics of Ara−1 show strong evidence of selective sweeps as well as clonal interference between competing lineages bearing different beneficial mutations. In some cases, sets of several mutations approached fixation simultaneously, often conveying no information about their order of origination; we present several possible explanations for the existence of these mutational cohorts. Against a backdrop of rapid selective sweeps both earlier and later, two genetically diverged clades coexisted for >6000 generations before one went extinct. In that time, many additional mutations arose in the clade that eventually prevailed. We show that the clades evolved a frequency-dependent interaction, which prevented the immediate competitive exclusion of either clade, but which collapsed as beneficial mutations accumulated in the clade that prevailed. Clonal interference and frequency dependence can occur even in the simplest microbial populations. Furthermore, frequency dependence may generate dynamics that extend the period of coexistence that would otherwise be sustained by clonal interference alone.},
keywords = {Demography and Ecology, Fitness Trajectories, Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
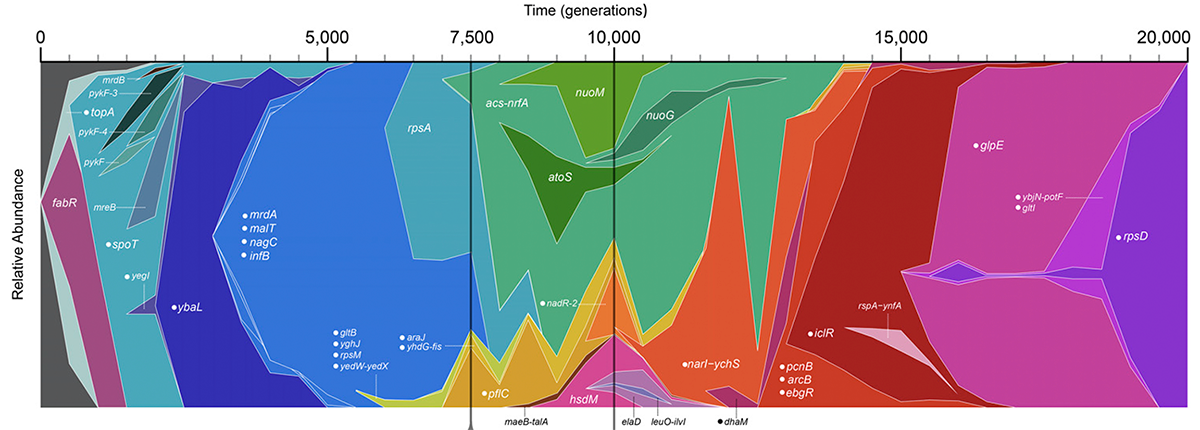
2014
Raeside C; Gaffé J; Deatherage D E; Tenaillon O; Briska A M; Ptashkin R N; Cruveiller S; Médigue C; Lenski R E; Barrick J E; Schneider D
Large Chromosomal Rearrangements during a Long-Term Evolution Experiment with Escherichia coli Journal Article
mBio, vol. 5, no. 5, pp. 1–13, 2014, ISSN: 2161-2129.
Abstract | Links | BibTeX | Altmetric | Tags: Genome Evolution
@article{Raeside2014,
title = {Large Chromosomal Rearrangements during a Long-Term Evolution Experiment with \textit{Escherichia coli}},
author = {Colin Raeside and Joël Gaffé and Daniel E. Deatherage and Olivier Tenaillon and Adam M. Briska and Ryan N. Ptashkin and Stéphane Cruveiller and Claudine Médigue and Richard E. Lenski and Jeffrey E. Barrick and Dominique Schneider},
editor = {Søren Molin and Fernando Baquero},
url = {https://journals.asm.org/doi/10.1128/mBio.01377-14},
doi = {10.1128/mBio.01377-14},
issn = {2161-2129},
year = {2014},
date = {2014-10-01},
urldate = {2014-10-01},
journal = {mBio},
volume = {5},
number = {5},
pages = {1--13},
abstract = {Large-scale rearrangements may be important in evolution because they can alter chromosome organization and gene expression in ways not possible through point mutations. In a long-term evolution experiment, twelve \textit{Escherichia coli} populations have been propagated in a glucose-limited environment for over 25 years. We used whole-genome mapping (optical mapping) combined with genome sequencing and PCR analysis to identify the large-scale chromosomal rearrangements in clones from each population after 40,000 generations. A total of 110 rearrangement events were detected, including 82 deletions, 19 inversions, and 9 duplications, with lineages having between 5 and 20 events. In three populations, successive rearrangements impacted particular regions. In five populations, rearrangements affected over a third of the chromosome. Most rearrangements involved recombination between insertion sequence (IS) elements, illustrating their importance in mediating genome plasticity. Two lines of evidence suggest that at least some of these rearrangements conferred higher fitness. First, parallel changes were observed across the independent populations, with ~65% of the rearrangements affecting the same loci in at least two populations. For example, the ribose-utilization operon and the \textit{manB} - \textit{cpsG} region were deleted in 12 and 10 populations, respectively, suggesting positive selection, and this inference was previously confirmed for the former case. Second, optical maps from clones sampled over time from one population showed that most rearrangements occurred early in the experiment, when fitness was increasing most rapidly. However, some rearrangements likely occur at high frequency and may have simply hitchhiked to fixation. In any case, large-scale rearrangements clearly influenced genomic evolution in these populations.},
keywords = {Genome Evolution},
pubstate = {published},
tppubtype = {article}
}
Quandt E M; Deatherage D E; Ellington A D; Georgiou G; Barrick J E
Recursive genomewide recombination and sequencing reveals a key refinement step in the evolution of a metabolic innovation in Escherichia coli Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 111, no. 6, pp. 2217–2222, 2014, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Citrate Evolution, Genotypes and Phenotypes, Methods and Miscellaneous
@article{Quandt2014,
title = {Recursive genomewide recombination and sequencing reveals a key refinement step in the evolution of a metabolic innovation in \textit{Escherichia coli}},
author = {Erik M. Quandt and Daniel E. Deatherage and Andrew D. Ellington and George Georgiou and Jeffrey E. Barrick},
url = {http://www.pnas.org/lookup/doi/10.1073/pnas.1314561111},
doi = {10.1073/pnas.1314561111},
issn = {0027-8424},
year = {2014},
date = {2014-02-01},
urldate = {2014-02-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {111},
number = {6},
pages = {2217--2222},
abstract = {Evolutionary innovations often arise from complex genetic and ecological interactions, which can make it challenging to understand retrospectively how a novel trait arose. In a long-term experiment, \textit{Escherichia coli} gained the ability to use abundant citrate (Cit+) in the growth medium after ~31,500 generations of evolution. Exploiting this previously untapped resource was highly beneficial: later Cit+ variants achieve a much higher population density in this environment. All Cit+ individuals share a mutation that activates aerobic expression of the \textit{citT} citrate transporter, but this mutation confers only an extremely weak Cit+ phenotype on its own. To determine which of the other >70 mutations in early Cit+ clones were needed to take full advantage of citrate, we developed a recursive genomewide recombination and sequencing method (REGRES) and performed genetic backcrosses to purge mutations not required for Cit+ from an evolved strain. We discovered a mutation that increased expression of the \textit{dctA} C4-dicarboxylate transporter greatly enhanced the Cit+ phenotype after it evolved. Surprisingly, strains containing just the \textit{citT} and \textit{dctA} mutations fully use citrate, indicating that earlier mutations thought to have potentiated the initial evolution of Cit+ are not required for expression of the refined version of this trait. Instead, this metabolic innovation may be contingent on a genetic background, and possibly ecological context, that enabled \textit{citT} mutants to persist among competitors long enough to obtain \textit{dctA} or equivalent mutations that conferred an overwhelming advantage. More generally, refinement of an emergent trait from a rudimentary form may be crucial to its evolutionary success.},
keywords = {Citrate Evolution, Genotypes and Phenotypes, Methods and Miscellaneous},
pubstate = {published},
tppubtype = {article}
}
2013
Barrick J E; Lenski R E
Genome dynamics during experimental evolution Journal Article
Nature Reviews Genetics, vol. 14, no. 12, pp. 827–839, 2013, ISSN: 1471-0056.
Abstract | Links | BibTeX | Altmetric | Tags: Review Articles
@article{Barrick2013,
title = {Genome dynamics during experimental evolution},
author = {Jeffrey E. Barrick and Richard E. Lenski},
url = {http://www.nature.com/articles/nrg3564},
doi = {10.1038/nrg3564},
issn = {1471-0056},
year = {2013},
date = {2013-12-01},
urldate = {2013-12-01},
journal = {Nature Reviews Genetics},
volume = {14},
number = {12},
pages = {827--839},
abstract = {Evolutionary changes in organismal traits may occur either gradually or suddenly. However, until recently, there has been little direct information about how phenotypic changes are related to the rate and the nature of the underlying genotypic changes. Technological advances that facilitate whole-genome and whole-population sequencing, coupled with experiments that 'watch' evolution in action, have brought new precision to and insights into studies of mutation rates and genome evolution. In this Review, we discuss the evolutionary forces and ecological processes that govern genome dynamics in various laboratory systems in the context of relevant population genetic theory, and we relate these findings to evolution in natural populations.},
keywords = {Review Articles},
pubstate = {published},
tppubtype = {article}
}
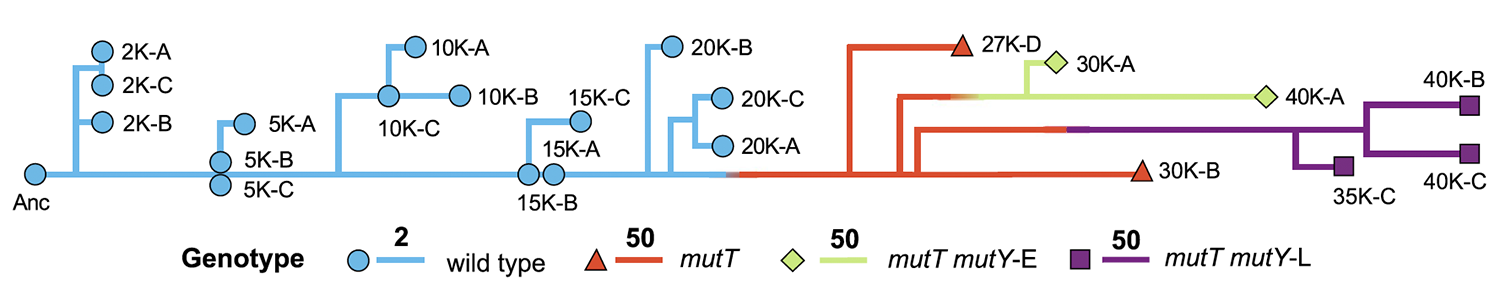
Wielgoss S; Barrick J E; Tenaillon O; Wiser M J; Dittmar W J; Cruveiller S; Chane-Woon-Ming B; Médigue C; Lenski R E; Schneider D
Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load Journal Article
Proceedings of the National Academy of Sciences of the United States of America, vol. 110, no. 1, pp. 222–227, 2013, ISSN: 0027-8424.
Abstract | Links | BibTeX | Altmetric | Tags: Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Theory and Simulations
@article{Wielgoss2013,
title = {Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load},
author = {Sébastien Wielgoss and Jeffrey E. Barrick and Olivier Tenaillon and Michael J. Wiser and W. James Dittmar and Stéphane Cruveiller and Béatrice Chane-Woon-Ming and Claudine Médigue and Richard E. Lenski and Dominique Schneider},
url = {http://www.pnas.org/cgi/doi/10.1073/pnas.1219574110},
doi = {10.1073/pnas.1219574110},
issn = {0027-8424},
year = {2013},
date = {2013-01-01},
urldate = {2013-01-01},
journal = {Proceedings of the National Academy of Sciences of the United States of America},
volume = {110},
number = {1},
pages = {222--227},
abstract = {Mutations are the ultimate source of heritable variation for evolution. Understanding how mutation rates themselves evolve is thus essential for quantitatively understanding many evolutionary processes. According to theory, mutation rates should be minimized for well-adapted populations living in stable environments, whereas hypermutators may evolve if conditions change. However, the long-term fate of hypermutators is unknown. Using a phylogenomic approach, we found that an adapting \textit{Escherichia coli} population that first evolved a \textit{mutT} hypermutator phenotype was later invaded by two independent lineages with \textit{mutY} mutations that reduced genome-wide mutation rates. Applying neutral theory to synonymous substitutions, we dated the emergence of these mutations and inferred that the \textit{mutT} mutation increased the point-mutation rate by ~150-fold, whereas the \textit{mutY} mutations reduced the rate by ~40-60%, with a corresponding decrease in the genetic load. Thus, the long-term fate of the hypermutators was governed by the selective advantage arising from a reduced mutation rate as the potential for further adaptation declined.},
keywords = {Fitness Trajectories, Genome Evolution, Genotypes and Phenotypes, Mutation Rates, Theory and Simulations},
pubstate = {published},
tppubtype = {article}
}